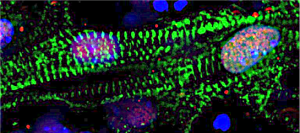
Researchers harness new pooled, image-based screening method to probe the functions of over 5,000 essential genes in human cells.
Nicole Davis | Whitehead Institute
November 21, 2022
A team of scientists at the Whitehead Institute for Biomedical Research and the Broad Institute of MIT and Harvard has systematically evaluated the functions of over 5,000 essential human genes using a novel, pooled, imaged-based screening method. Their analysis harnesses CRISPR-Cas9 to knock out gene activity and forms a first-of-its-kind resource for understanding and visualizing gene function in a wide range of cellular processes with both spatial and temporal resolution. The team’s findings span over 31 million individual cells and include quantitative data on hundreds of different parameters that enable predictions about how genes work and operate together. The new study appears in the Nov. 7 online issue of the journal Cell.
“For my entire career, I’ve wanted to see what happens in cells when the function of an essential gene is eliminated,” says MIT Professor Iain Cheeseman, who is a senior author of the study and a member of Whitehead Institute. “Now, we can do that, not just for one gene but for every single gene that matters for a human cell dividing in a dish, and it’s enormously powerful. The resource we’ve created will benefit not just our own lab, but labs around the world.”
Systematically disrupting the function of essential genes is not a new concept, but conventional methods have been limited by various factors, including cost, feasibility, and the ability to fully eliminate the activity of essential genes. Cheeseman, who is the Herman and Margaret Sokol Professor of Biology at MIT, and his colleagues collaborated with MIT Associate Professor Paul Blainey and his team at the Broad Institute to define and realize this ambitious joint goal. The Broad Institute researchers have pioneered a new genetic screening technology that marries two approaches — large-scale, pooled, genetic screens using CRISPR-Cas9 and imaging of cells to reveal both quantitative and qualitative differences. Moreover, the method is inexpensive compared to other methods and is practiced using commercially available equipment.
“We are proud to show the incredible resolution of cellular processes that are accessible with low-cost imaging assays in partnership with Iain’s lab at the Whitehead Institute,” says Blainey, a senior author of the study, an associate professor in the Department of Biological Engineering at MIT, a member of the Koch Institute for Integrative Cancer Research at MIT, and a core institute member at the Broad Institute. “And it’s clear that this is just the tip of the iceberg for our approach. The ability to relate genetic perturbations based on even more detailed phenotypic readouts is imperative, and now accessible, for many areas of research going forward.”
Cheeseman adds, “The ability to do pooled cell biological screening just fundamentally changes the game. You have two cells sitting next to each other and so your ability to make statistically significant calculations about whether they are the same or not is just so much higher, and you can discern very small differences.”
Cheeseman, Blainey, lead authors Luke Funk and Kuan-Chung Su, and their colleagues evaluated the functions of 5,072 essential genes in a human cell line. They analyzed four markers across the cells in their screen — DNA; the DNA damage response, a key cellular pathway that detects and responds to damaged DNA; and two important structural proteins, actin and tubulin. In addition to their primary screen, the scientists also conducted a smaller, follow-up screen focused on some 200 genes involved in cell division (also called “mitosis”). The genes were identified in their initial screen as playing a clear role in mitosis but had not been previously associated with the process. These data, which are made available via a companion website, provide a resource for other scientists to investigate the functions of genes they are interested in.
“There’s a huge amount of information that we collected on these cells. For example, for the cells’ nucleus, it is not just how brightly stained it is, but how large is it, how round is it, are the edges smooth or bumpy?” says Cheeseman. “A computer really can extract a wealth of spatial information.”
Flowing from this rich, multi-dimensional data, the scientists’ work provides a kind of cell biological “fingerprint” for each gene analyzed in the screen. Using sophisticated computational clustering strategies, the researchers can compare these fingerprints to each other and construct potential regulatory relationships among genes. Because the team’s data confirms multiple relationships that are already known, it can be used to confidently make predictions about genes whose functions and/or interactions with other genes are unknown.
There are a multitude of notable discoveries to emerge from the researchers’ screening data, including a surprising one related to ion channels. Two genes, AQP7 and ATP1A1, were identified for their roles in mitosis, specifically the proper segregation of chromosomes. These genes encode membrane-bound proteins that transport ions into and out of the cell. “In all the years I’ve been working on mitosis, I never imagined ion channels were involved,” says Cheeseman.
He adds, “We’re really just scratching the surface of what can be unearthed from our data. We hope many others will not only benefit from — but also build upon — this resource.”
This work was supported by grants from the U.S. National Institutes of Health as well as support from the Gordon and Betty Moore Foundation, a National Defense Science and Engineering Graduate Fellowship, and a Natural Sciences and Engineering Research Council Fellowship.