Raleigh McElvery
February 26, 2020
After decades of speculation, researchers have demonstrated that a classical DNA repair enzyme also binds to RNA, affecting blood cell development.
The DNA-dependent protein kinase, otherwise known as DNA-PK, is one of the most important enzymes that binds DNA and repairs double-stranded breaks. This mode of repair is essential for generating receptors that help the immune system fight off intruders. But DNA-PK doesn’t just bind DNA; it also binds RNA. Although researchers have known this for decades, they didn’t fully understand what kinds of RNAs DNA-PK bound in mammalian cells, or the physiological consequences of this binding.
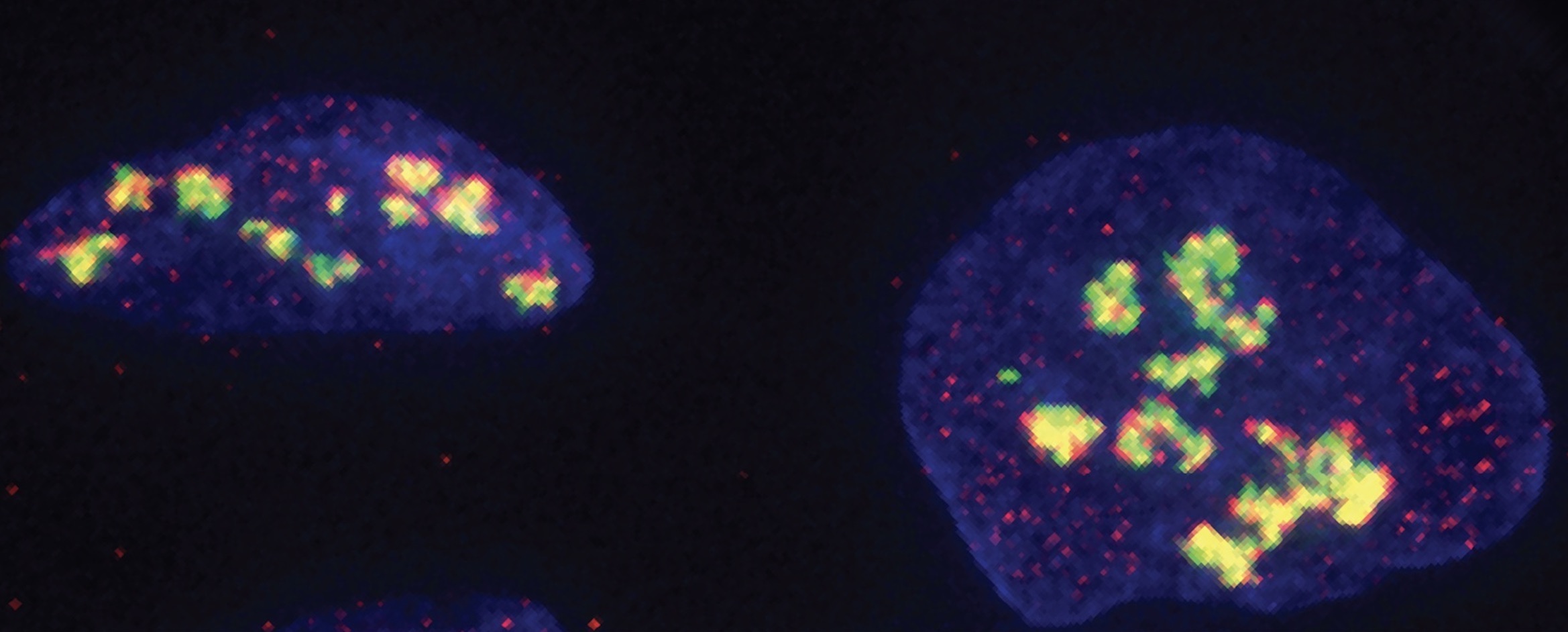
In a new study published on February 26 in Nature, researchers from MIT and Columbia University have uncovered a mechanism whereby DNA-PK binds to the RNA involved in ribosome assembly. Ribosomes — the cell’s protein synthesis machinery — ensure that stem cells give rise to enough red blood cells. The researchers found that mutating DNA-PK prevents the ribosomes from being built properly, which prevents blood cells from doing their job and leads to blood disorders.
“This is the first biochemical evidence of DNA-PK assembly and activation by RNA inside cells,” says Eliezer Calo, a co-senior author and assistant professor in MIT’s Department of Biology. “We’re still trying to determine the mechanisms that regulate protein synthesis in stem cells, and this study reveals one of them.”
Co-senior author, Shan Zha from Columbia University, had previously studied DNA-PK’s role in DNA repair by generating a mouse model that carried enzymatically-dead versions of DNA-PK. While using this model to investigate tumorigenesis, Zha’s lab found these mutant mice developed a form of blood cancer known as myeloid disease. At the same time, another research group showed that mutations in DNA-PK also led to anemia, which occurs when the body does not have enough healthy red blood cells
Neither myeloid disease nor anemia could be easily explained by DNA repair defects alone. However, the two blood disorders did share some similarities to diseases caused by ribosome defects. Because DNA-PK resides in the same organelle where ribosomes are made, the Zha and Calo labs began to wonder whether DNA-PK could bind to the RNA there and control ribosome biogenesis.
In this new study, the Zha lab found that DNA-PK mutations impaired protein translation in red blood cell progenitors, which might contribute to anemia. In parallel, the Calo lab was investigating ribosomal RNA processing and was surprised to find that DNA-PK seemed to be implicated in ribosome assembly. The Calo lab then mapped all the RNAs in cells that bind DNA-PK. The enzyme unexpectedly attached to U3, a small RNA that helps assemble one of the subunits comprising the ribosome. Once it binds U3, DNA-PK can transfer a phosphate group to several specific sites on one of its own subunits. If DNA-PK is defective and cannot transfer the phosphate group, protein synthesis in blood stem cells is impaired, eventually causing anemia.
DNA-PK is essential for cellular viability in nearly all human cell lines, including cancer cell lines, while many other proteins involved in same DNA repair pathway are dispensable. Several studies, including one published by the Zha lab, showed that DNA-PK protein levels are 50-fold higher in common human cell lines than in rodent cell lines. The researchers do not yet know why the enzyme is so critical, but they suspect it might have to do with its ability to bind RNA. “We are interested in exploring whether this new role for DNA-PK could provide clues to this puzzle,” Zha says.
Calo says their findings could also have important implications for cancer treatment, because DNA-PK has emerged as a promising target for cancer therapy. Drugs that inhibit DNA-PK could prevent cancer cells from repairing their DNA and replicating successfully, but he warns these same remedies could also impact stem cell function. The next step is to explore DNA-PK’s other RNA binding targets and the related molecular pathways.
“We’ve demonstrated that DNA-PK has an entirely separate role that has nothing to do with DNA repair,” Calo says. “In the future, we’re excited to learn what additional RNA-related duties it may have beyond stem cell maintenance.”
Top Image: Ribosomes are assembled in the nucleoli (shown here in human cells).
Citation:
“DNA-PKcs has KU-dependent function in rRNA processing and haematopoiesis”
Nature, online February 26, 2020, DOI: 10.1038/s41586-020-2041-2
Zhengping Shao, Ryan A. Flynn, Jennifer L. Crowe, Yimeng Zhu, Jialiang Liang, Wenxia Jiang, Fardin Aryan, Patrick Aoude, Carolyn R. Bertozzi, Verna M. Estes, Brian J. Lee, Govind Bhagat, Shan Zha, and Eliezer Calo