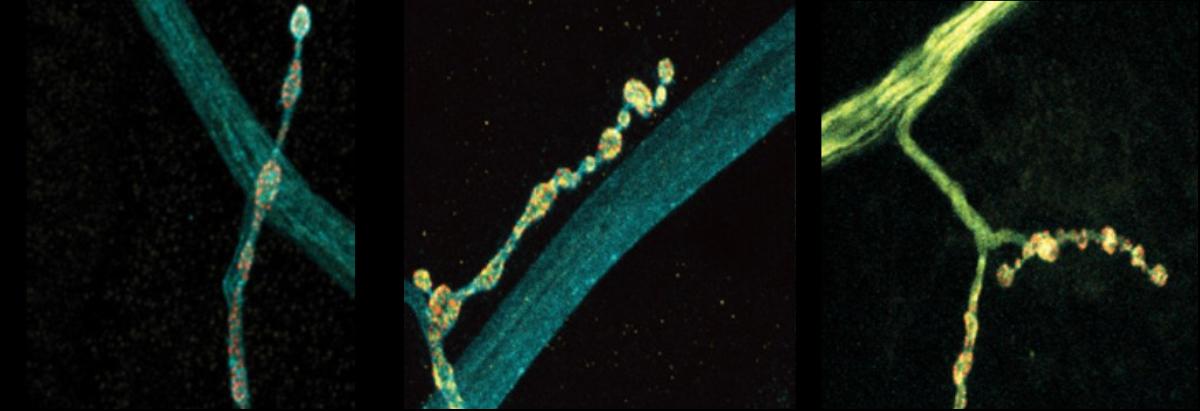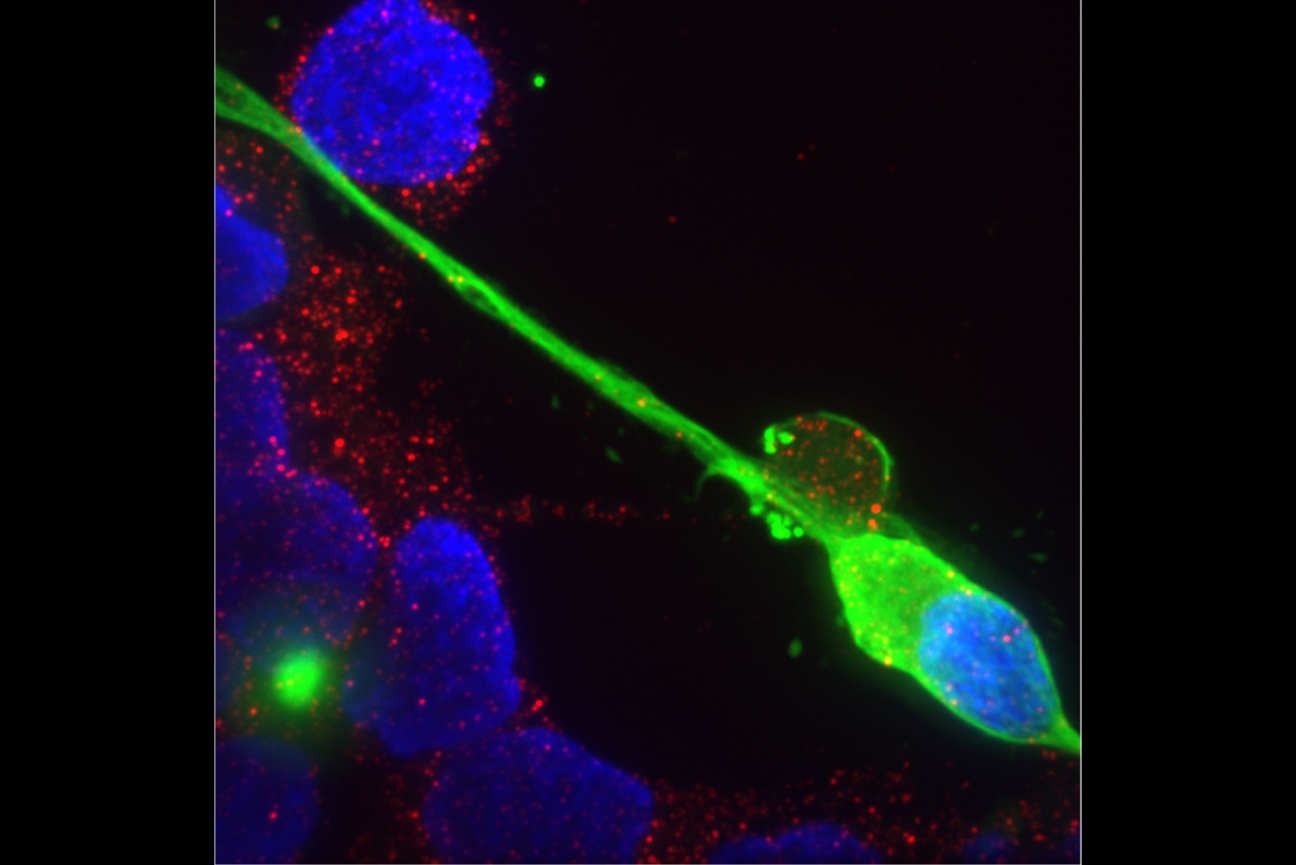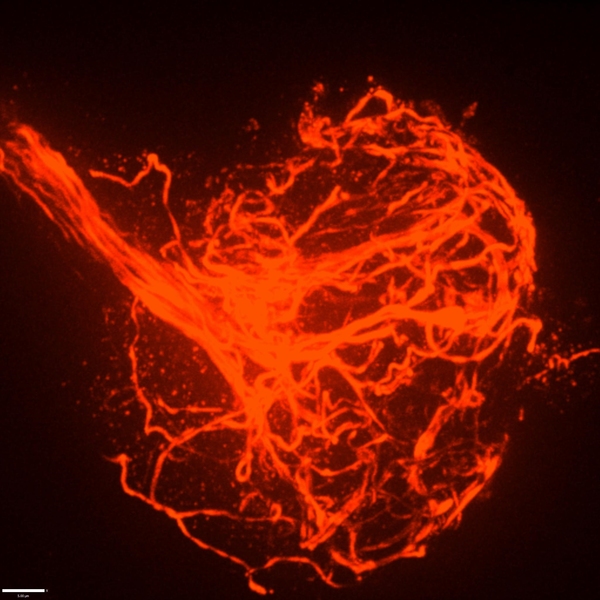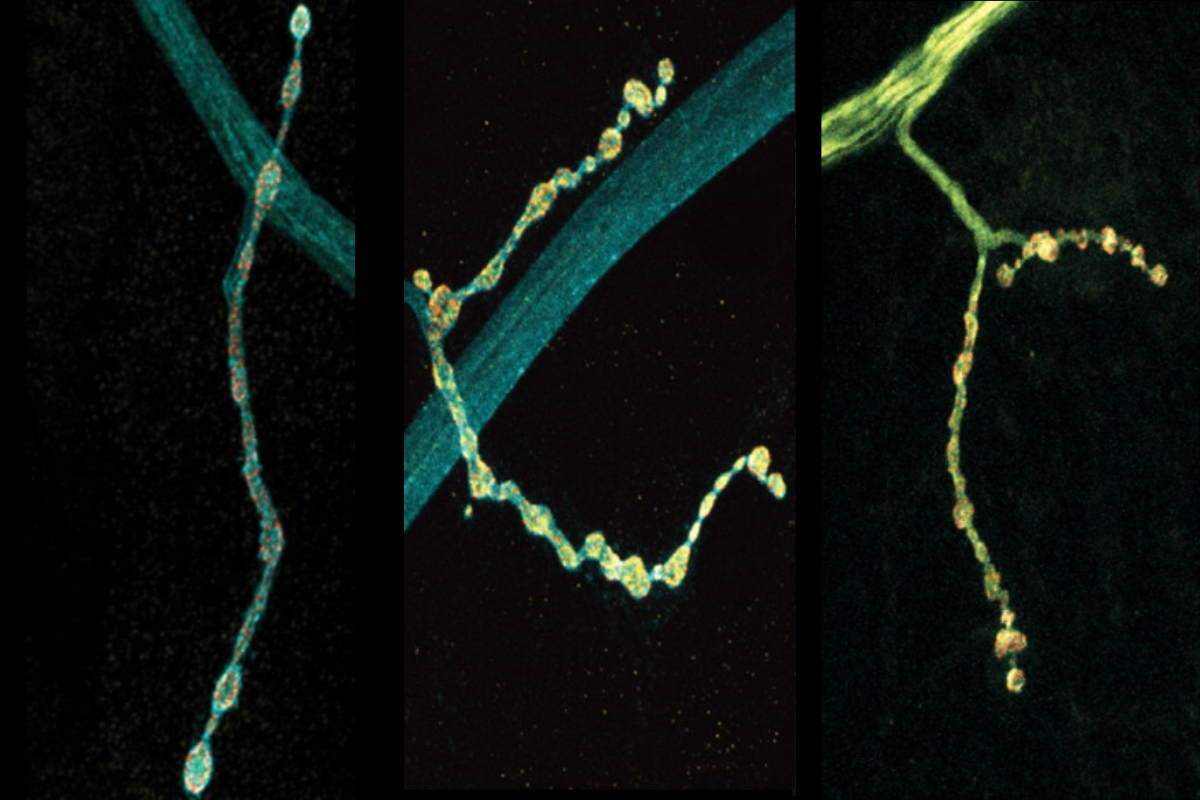
Neurons stochastically generated up to eight different versions of a protein-regulating neurotransmitter release, which could vary how they communicate with other cells.
David Orenstein | The Picower Institute for Learning and Memory
September 25, 2023
Neurons are talkers. They each communicate with fellow neurons, muscles, or other cells by releasing neurotransmitter chemicals at “synapse” junctions, ultimately producing functions ranging from emotions to motions. But even neurons of the exact same type can vary in their conversational style. A new open-access study in Cell Reports by neurobiologists at The Picower Institute for Learning and Memory highlights a molecular mechanism that might help account for the nuanced diversity of neural discourse.
The scientists made their findings in neurons that control muscles in Drosophila fruit flies. These cells are models in neuroscience because they exhibit many fundamental properties common to neurons in people and other animals, including communication via the release of the neurotransmitter glutamate. In the lab of Troy Littleton, Menicon Professor in MIT’s departments of Biology and Brain and Cognitive Sciences, which studies how neurons regulate this critical process, researchers frequently see that individual neurons vary in their release patterns. Some “talk” more than others.
In more than a decade of studies, Littleton’s lab has shown that a protein called complexin has the job of restraining spontaneous glutamate chatter. It clamps down on fusion of glutamate-filled vesicles at the synaptic membrane to preserve a supply of the neurotransmitter for when the neuron needs it for a functional reason, for instance to simulate a muscle to move. The lab’s studies have identified two different kinds of complexin in flies (mammals have four) and showed that the clamping effectiveness of the rare but potent 7B splice form is regulated by a molecular process called phosphorylation. How the much more abundant 7A version is regulated was not known, but scientists had shown that the RNA transcribed from DNA that instructs the formation of the protein is sometimes edited in the cell by an enzyme called ADAR.
In the new study from Littleton’s team, led by Elizabeth Brija PhD ’23, the lab investigated whether RNA editing of complexin 7A affects how it regulates glutamate release. What she discovered was surprising. Not only does RNA editing of complexin 7A have a significant impact on how well the protein prevents glutamate release, but also this can vary widely among individual neurons because they can stochastically mix and match up to eight different editions of the protein. Some edits were much more common than others on average, but 96 percent of the 200 neurons the team examined had at least some editing, which affected the structure of an end of the protein called its C-terminus. Experiments to test some of the consequences of this structural variation showed that different complexin 7A edits can dramatically affect the level of electrical current measurable at different synapses. That varying level of activity can also affect the growth of the synapses the neurons make with muscle. RNA editing of the protein might therefore endow each neuron with fine degrees of communication control.
“What this offers the nervous system is that you can take the same transcriptome and by alternatively editing various RNA transcripts, these neurons will behave differently,” Littleton says.
Moreover, Littleton and Brija’s team found that other key proteins involved in synaptic glutamate release, such as synapsin and Syx1A, are also sometimes edited at quite different levels among the same population of neurons. This suggests that other aspects of synaptic communication might also be tunable.
“Such a mechanism would be a robust way to change multiple features of neuronal output,” Brija, Littleton, and colleagues wrote.
The team tracked the different editing levels by meticulously extracting and sequencing RNA from the nuclei and cell bodies of 200 motor neurons. The work yielded a rich enough dataset to show that any of three adenosine nucleotides encoding two amino acids in the C-terminus could be swapped for another, yielding eight different editions of the protein. A slim majority of complexin 7A went unedited in the average neuron, while the seven edited versions composed the rest with widely varying degrees of frequency.
To investigate the functional consequences of some of the different editions, the team knocked out complexin and then “rescued” flies by adding back in unedited or two different edited versions. The experiments showed a stark contrast between the two edited proteins. One, which occurs more commonly, proved to be a less effective clamp than unedited complexin, barely preventing spontaneous glutamate release and upticks in electrical current. The other turned out to be more effective at clamping than the unedited version, keeping a tight lid on glutamate release and synaptic output. And while both of the edited versions showed a tendency to drift away from synapses and into the neuron’s axon, the long branch that extends from the cell body, the edition that clamped well prevented any overgrowth of synapses while the one that clamped poorly provided only a meager curb.
Because multiple editions are often present in neurons, Brija and the team did one more set of experiments in which they “rescued” complexin-less flies with a combination of unedited complexin and the weak-clamping edition. The result was a blend of the two: reduced spontaneous glutamate release than with just the weakly clamping edition alone. The findings suggest that not only does each edition potentially fine-tune glutamate release, but that combinations among them can act in a combinatorial fashion.
In addition to Brija and Littleton the paper’s other authors are Zhuo Guan and Suresh Jetti.
The National Institutes of Health, The JPB Foundation, and The Picower Institute for Learning and Memory supported the research.