Graduate student Julie Monda has spent five years investigating two divergent aspects of cell division, revealing some unexpected results and new research questions.
Raleigh McElvery
July 23, 2018
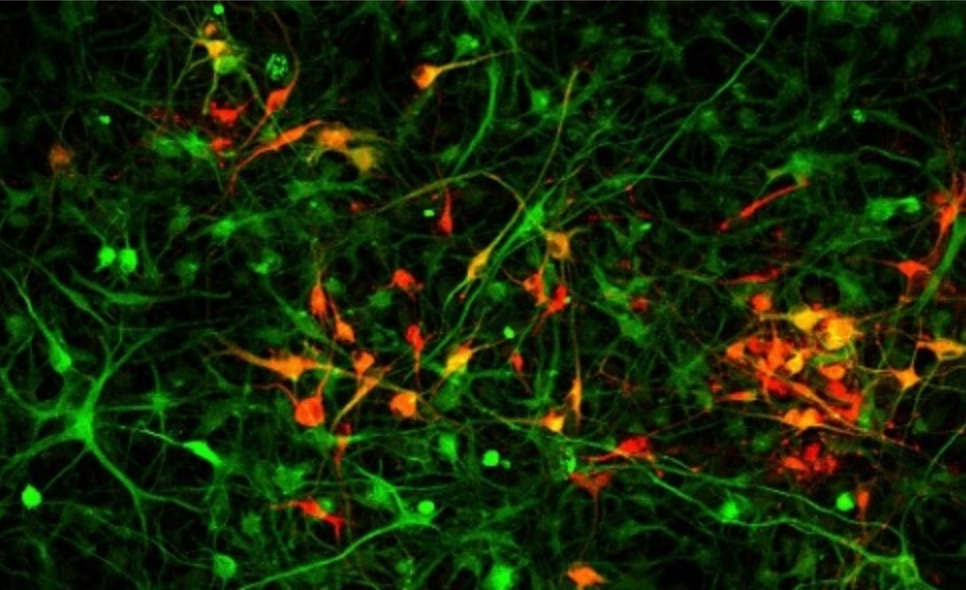
To sixth year graduate student Julie Monda, dividing cells are among the most beautiful things she’s ever seen. Watching the tiny, delicate spheres split into identical versions of themselves also provides her with a visual readout for her experiments — will the process continue if she removes a certain piece of a certain protein? Will the genetic material still distribute equally between the two cells? Which molecules are crucial for cell division, and how are they regulated?
Our cells are constantly dividing in order to grow and repair themselves, although some (like skin cells) do so more often than others, say, in the brain. This process, known as mitosis, is the primary focus of Iain Cheeseman’s lab, situated in the Whitehead Institute for Biomedical Research. Most of the research in the Cheeseman lab involves the kinetochore, a group of proteins located on the chromosome where the arms join. During mitosis, long, fibrous structures, known as microtubules, attach to the kinetochore to pull apart the duplicated chromosomes as the parent cell splits in half, ensuring each daughter cell receives an exact copy of the parent’s genetic blueprint.
Before she arrived at MIT Biology in the fall of 2012, Monda worked as a research technician at St. Jude Children’s Research Hospital in Memphis, Tennessee in the lab of Brenda Schulman PhD ’96 . As she recalls, she always “preferred performing hands-on research techniques at the lab bench over being in a classroom.” So she surprised even herself when she chose MIT’s graduate program in biology precisely because it requires all first-year students to take a full course load their fall semester before beginning lab rotations.
“That structure seemed useful given that I studied biochemistry as an undergraduate at the University of Tulsa, and the degree requirements were weighted more towards chemistry than biology,” she says. “Plus, when you’re only taking classes, you spend more time interacting with your classmates. It creates a close-knit community that extends throughout your entire graduate career and beyond.”
Monda ultimately selected the Cheeseman lab because it married her interests in biochemistry and cell biology.
“The research in this lab focuses on various elements of kinetochore function and cell division, but everyone is generally working on their own distinct questions,” she explains. “I knew I would have an area that was mine to explore. It’s both exciting and challenging because no one else is thinking about your projects to the extent that you are.”
Monda’s story is a tale of two projects: one focused on the interface between the kinetochore and the array of microtubules known as the mitotic “spindle,” and another project that ended up taking both her and the lab in a slightly new direction.
The first, concerning kinetochore-microtubule interactions, represented a collaboration with former lab technician Ian Whitney. For this endeavor, Monda investigated a protein complex called Ska1, found at the outer kinetochore.
The Ska1 complex is located where the kinetochore and microtubule meet. Ska1’s role, Monda explains, is to allow the kinetochores to remain attached to the spindle during chromosome segregation, even as the microtubules that compose the spindle begin to disassemble (as they must do).
“We wanted to know how the kinetochore hangs onto this polymer that is essentially falling apart,” Monda explains. “Long story short, we ended up defining specific surfaces within the Ska1 complex that are important for holding on to the microtubule as it shrinks, and — as we were surprised to note — also as it grows”
Although Ska1 only requires a single point of contact to bind a microtubule, Monda and Whitney pinpointed multiple surfaces on Ska1 that are required to allow it to remain associated with the microtubules as they disassemble and reassemble themselves.
While her Ska1 project was very much in line with the types of questions that the Cheeseman lab traditionally pursues, Monda also worked on another endeavor that “began as a side project and slowly evolved into a more full-time effort.” This project involves a motor protein called dynein, which helps to align the chromosomes and position the spindle during mitosis.
Dynein piqued Monda’s interest because of its role in mitosis, as well as its importance throughout the entire cell cycle. Motor proteins are molecules powered by the release of chemical energy that move along surfaces, sometimes transporting cargo, sometimes performing other essential tasks. Dynein is a motor protein that walks in one direction along microtubules, even when the microtubules latch onto the kinetochore to yank apart the chromosomes during mitosis.
But dynein doesn’t act alone. There are a number of additional proteins that also play a key role in coordinating its activity and localization. Monda is studying two of these accessory regulatory proteins, Nde1 and NdeL1, which bind to dynein and help promote some of its functions. She wanted to understand how Nde1 and NdeL1 interact with dynein to activate it. Although Nde1 and NdeL1 are nearly identical in function, Monda discovered that Nde1 (but not NdeL1) binds to another complex: the 26S proteasome.
The proteasome degrades proteins within the cell, influencing virtually all aspects of cellular function, including DNA synthesis and repair, transcription, translation, and cell signaling. Given its ubiquity, it has remained a point of interest among the scientific community for years. And yet, before Monda’s research, the interaction between Nde1 and the proteasome had apparently gone unnoticed. Researchers have long studied Nde1 in relation to dynein, but it’s possible that the interaction between Nde1 and the proteasome represents a new function for Nde1 unrelated to dynein regulation. In fact, Monda’s finding may have implications for understanding the development of the human brain.
“It’s clear that patients with mutations in Nde1 have much more severe neurodevelopmental defects than scientists would have predicted,” Monda says, “so it’s possible that this new interaction between Nde1 and the proteasome could help to explain why Nde1 is so important in the brain.”
Her most recent results have been published in Molecular Biology of the Cell.
“I’ve found some exciting results over the past few years,” Monda says, “and even though a lot of my research has gone in a direction that’s not strictly mitosis-related, Iain has been great about allowing me to follow the science wherever it leads. We want to know what these proteins are actually doing, both in terms of this new interaction and also more broadly within the cell.”
Monda intends to submit and defend her thesis this summer, and assume a postdoctoral position at the University California, San Diego in the fall. Although she’s been watching cells divide for years now, the process still retains its grandeur.
“Often times biologists investigate questions at scales where we can’t really see what we’re studying as we study it,” she says. “But having this visual readout makes it more tangible; I feel like I can better appreciate what exactly it is that I’m trying to understand, as well as the beauty and complexity of the processes that sustain life.”