Stem cells are the versatile building blocks from which every cell type in the body, from neurons, to skin cells, to blood cells, is ultimately descended. Researchers have also figured out how to turn stem cells into different cell types in the lab, which has been helpful for studying health and disease in their normal cellular contexts, and could be used to generate cells for medical transplants. Whitehead Institute Founding Member Rudolf Jaenisch not only uses these cells in his research, but has spent much of his career discovering and improving the methods for making accurate laboratory models out of stem cell-derived cells.
One challenge that Jaenisch’s lab is focusing on is how to eliminate the differences between cell types as they are found in the body and their stem cell-derived equivalents. In particular, they have found that stem-cell derived cells are often immature, more closely resembling the cells found in fetuses rather than in adults. These differences can make the cells less accurate research models and prevent them from being medically useful as functional transplant cells. Stem cells in the body receive complex cocktails of molecular signals as they transform into different cell types. The challenge for researchers lies in figuring out which of the many molecular signals in the body are relevant and then get the recipe exactly right in their recreations.
Postdoc Haiting Ma in the Jaenisch lab decided to tackle this problem for hepatocytes, the main type of cell in the liver. In work published in Cell Stem Cell on April 21, Jaenisch and Ma share their findings on why stem cell-derived liver cells resemble fetal liver cells, and what’s needed to make them mature—including an important role for a thyroid hormone.
The liver filters everything that enters the body through the digestive system. It helps to store and modify nutrients, safely break down toxins and waste, process medications, and more. There is still a lot to learn about how the liver functions, and what goes wrong in a number of liver-associated diseases, and accurate stem cell-derived models will help with that research. Liver cells are also needed to treat end-stage liver disease, and if researchers could mass produce stem cell-derived liver cells that can function safely in an adult liver, this could help to meet the demand for liver cell transfusions.
For this study, Jaenisch and Ma grew liver cells from stem cells in two setups: a typical 2D culture, in which the cells were grown in a dish, and a 3D spheroid, in which cells that started out in the normal culture were then allowed to grow into three-dimensional balls of cells. The spheroids can be designed to mimic some aspects of the cells’ natural environment in ways that a 2D culture cannot. In each case, the researchers exposed the cells to a carefully timed mixture of signals to prompt them to develop into liver cells. The researchers then analyzed cells from both the 2D and 3D cultures and compared them to primary liver cells, or cells from a body, using a variety of techniques to look for differences related to DNA and gene expression. They found that the cells cultivated in the 3D system were closer to cells from the adult body than those in the 2D system.
“The 3D culture not only contributes to maturation of the liver cells, but it can also be used to scale up production of the cells, which could be very useful for cell therapies in the future,” Ma says.
However, both sets of lab-derived cells lacked important features of adult liver cells. The analyses pointed to one important missing factor in particular: in the adult liver cells, a hormone receptor called Thyroid Hormone Receptor Beta (THRB) binds to a number of places in the DNA. THRB then senses the presence or absence of thyroid hormones, and regulates a variety of gene expression processes accordingly. However, the researchers found that while the stem cell-derived liver cells made the right amount of THRB, something was preventing it from binding where it should and performing its function.
Normally, THRB has a partner that helps it bind to DNA, the thyroid hormone T3. When the researchers added T3 to their 2D and 3D cultures, this led to more typical binding of THRB, which in turn made the cells—especially the cells from the 3D culture—more closely resemble adult liver cells in a number of ways. Improved THRB binding increased the expression of key liver genes, restored the activity of regulatory elements in the DNA that modify gene expression, and reduced the expression of a fetal liver gene. The researchers also gained insights into the molecules that THRB interacts with and the mechanisms by which it affects liver maturation, painting a more complete picture of its key roles in liver cells.
Altogether, this work led to a better recipe for making adult liver cells from stem cells in the lab–using the 3D spheroid culture and adding T3. When cells developed with this approach were incorporated into the livers of mice, the cells integrated successfully and the liver maintained normal function long term.
The new and improved stem cell-derived liver cells are still not a perfect match for adult liver cells—the researchers have ideas about which missing characteristics they could tackle next—but the current cells’ ability to seamlessly integrate into the liver, as well as indicators from the analyses that they would be good models for liver-associated diseases, suggest that they will be useful in a variety of projects.
“As we improve the authenticity of our stem cell-derived cell types, we open up new opportunities for research,” Jaenisch says. “We can build more accurate models in which to study high-impact diseases, such as liver diseases, diabetes, and chronic viral infections, and using those models we can develop strategies for treatment and prevention.”
Merrill Meadow | Whitehead Institute
April 20, 2022
This year’s Vanderbilt Prize in Biomedical Science will be awarded to Whitehead Institute director Ruth Lehmann. It recognizes women scientists with a stellar record of research accomplishments who also have made significant contributions to mentoring other women in science.
“Dr. Lehmann’s determination to solve the deepest mysteries of life while encouraging others at the beginning of their careers exemplifies the spirit of the Vanderbilt Prize in Biomedical Science,” says Jeff Balser, president and chief executive officer of Vanderbilt University Medical Center – which bestows the Prize – and dean of Vanderbilt University School of Medicine. “I’m honored to congratulate her as the 2022 Vanderbilt Prize recipient.”
“I’m thrilled to be receiving this honor, recognizing the importance of mentoring and empowering the next generation of scientists,” says Lehmann, who has mentored scores of students and research fellows during her career, and has developed a mentorship program specifically designed to encourage and empower junior faculty in science. Read more here.
Graduate student En Ze Linda Zhong-Johnson is creating new methods to measure and enhance enzyme activity — which she hopes will help restore a plastic-choked world.
Grace van Deelen
April 21, 2022
After graduating with her undergraduate degree in molecular genetics from the University of Toronto in 2016, En Ze Linda Zhong-Johnson celebrated with a trip to Alaska. There, she saw a pristine landscape unlike the plastic-littered shores of the Toronto waterfront. “What I saw up there was so different from what I saw in the city,” Zhong-Johnson says. “I realized there shouldn’t be all this waste floating everywhere, in our water, in our environment. It’s not natural.”
As a trained biologist, Zhong-Johnson began to think about the problem of plastic pollution from a biological perspective. One solution, she thought, could be biological recycling: a process by which living organisms break down materials, using digestion or other metabolic processes to turn these materials into smaller pieces or new compounds. Composting, for example, is a type of biological recycling — microbes in the soil break down discarded food, speeding up the decomposition process. Zhong-Johnson wondered if there were any organisms on Earth that could use the carbon in polyethylene terephthalate (PET), a common plastic used in water bottle and food packaging, as an energy source.
Earlier that same year, Japanese scientists discovered that a bacterium, Ideonella sakaiensis, could do just that by producing enzymes that could break down PET. The two main PET-degrading enzymes, referred to as IsPETase and IsMHETase, are able to turn PET into two chemical compounds, terephthalic acid and ethylene glycol, which I. sakaiensis can use for food.
The discovery of these enzymes opened up many new questions and possible applications that scientists have continued to work on since. However, because there was — and still is — much to learn about PET-degrading enzymes, they are still not widely used to recycle consumer products. Zhong-Johnson figured that, in graduate school, she could build on the existing IsPETase research and help to accelerate their use at recycling facilities. Specifically, she wanted to engineer the enzyme to work faster at lower temperatures, and study how, fundamentally, the enzymes worked on the surface of PET plastic to degrade it.
“I hopped on the excitement train, along with the rest of the world,” she says.
A better enzyme
After receiving her acceptance to MIT to complete her PhD, Zhong-Johnson approached various professors, pitching her idea to speed up IsPETase activity. Christopher Voigt, the Daniel I. C. Wang Professor of Biological Engineering, and Anthony Sinskey, professor of biology, were interested, and formed a co-advisorship to support Zhong-Johnson’s project. Sinskey, in particular, was impressed by her idea to help solve the world’s plastic problem with PET-degrading enzymes.
Zhong-Johnson screens for enzyme variants with improved activity. Credit: Grace van Deelen
“Plastic pollution is a big problem,” he says, “and that’s the kind of problem my lab likes to tackle.” Plus, he says, he feels “committed to helping graduate students who want to apply their science and technology learnings to the environment.”
While the idea of a plastic-degrading enzyme seems like a panacea, the enzyme’s practical applications have been limited by its biology. The wild-type IsPETase is a mesophilic enzyme, meaning the structure of the enzyme is only stable around ambient temperatures, and the enzyme loses its activity above that threshold. This restriction on temperature limits the number and types of facilities that can use IsPETase, as well as the rate of the enzyme reaction, and drives up the cost of their use.
However, Zhong-Johnson thinks that, with combined approaches of biological and chemical engineering, it’s possible to scale up the use of the enzymes by increasing their stability and activity. For example, an enzyme that’s highly active at lower temperatures could work in unheated facilities, or even be sprinkled directly into landfills or oceans to degrade plastic waste — a process called bioremediation. Increasing the activity of the enzyme at ambient temperatures could also expand the possible applications.
“Most of the environments where plastic is present are not above 50 degrees Celsius,” said Zhong-Johnson. “If we can increase enzyme activity at lower temperatures, that’s really interesting for bioremediation purposes.”
Now a fifth-year graduate student, Zhong-Johnson has honed her project, and is focusing on increasing the activity of IsPETase. To do so, she’s using directed evolution — creating random mutations in the IsPETase gene, and selecting for IsPETase variants that digest PET faster. When they do, she combines the beneficial mutations and uses that as template for the next round of library generation, to improve the enzyme even further. The evolution is “directed” because Zhong-Johnson herself, rather than nature, is picking out which gene sequences of enzyme proceed through to the next round of random mutagenesis, and which don’t. Her ultimate goal is to create a more efficient and hardier enzyme that will, hopefully, work faster at ambient temperatures.
A better protocol
Just as Zhong-Johnson was beginning her project, she ran into an obstacle: There wasn’t a standard way to measure whether her experiments were successful. In particular, no immediately applicable method existed to measure enzyme kinetics for IsPETases on solid substrates like plastic bottles and other plasticware. That was a problem for Zhong-Johnson because understanding enzyme activity was a crucial part of how she selected her enzymes in the directed evolution process.
Usually, enzyme activity is measured via product accumulation: When enzymes metabolize a substance, they create a new substance in return, called a product. Measuring the amount of product created by an enzyme after a certain amount of time gives the researcher a snapshot of that enzyme’s activity.
There are two problems with the product accumulation method, though. First, it is usually done using liquid or soluble substrates. In other words, the material that the enzyme is targeting is dissolved, like sugar dissolved in water. Then, the enzyme is added to that liquid concoction and mixed evenly throughout. However, the substrate Zhong-Johnson wanted to use — PET — was not soluble but solid, meaning it could not be evenly distributed like a soluble substrate. Second, the product accumulation measurement methods available were only practical for measuring less than a handful of timepoints for a few enzyme or substrate concentrations. As a result, many in the field opted to measure a single time point, late in the enzyme reaction, which doesn’t provide an indication of how an enzyme’s rate of digestion actually changes over the course of time — something that can be measured through kinetic measurements.
Taking kinetic measurements would help researchers like Zhong-Johnson illustrate the full pattern of enzyme activity and answer questions like: When is the enzyme most active? Does most product accumulation happen at the beginning of the reaction or the end? How does temperature impact the rate of these reactions over time? To answer these questions, she realized she would have to develop the method herself.
Through a serendipitous discussion with a group of chemical engineering undergraduate students that Zhong-Johnson was mentoring, she came up with a solution, which she published in a 2021 paper in Scientific Reports. The undergraduates brought to her attention many factors that she had overlooked about the enzyme, and she says she would not have realized the importance of kinetic measurements if it weren’t for the fact that she was trying to design an experiment that the undergraduates could perform over the course of three hours.
The paper outlined a new way to measure enzyme activity, which Zhong-Johnson calls “the bulk absorbance method.” Instead of measuring the final product accumulation at very late time points, the bulk absorbance method involves taking multiple kinetic measurements at early time intervals during the experiment. This technique informs Zhong-Johnson’s directed evolution approach: If she can find which enzymes are most active at low temperatures, she can select the best possible enzyme for the next round of analyses. She hasn’t yet engineered an enzyme she’s completely happy with, but she’s gotten much closer to her ultimate goal.
Solving big problems together
Zhong-Johnson’s discoveries have been made possible by the collaboration between her and her two co-advisors, Voigt and Sinskey, who have supported her independence throughout her five years at MIT.
Zhong-Johnson and her advisor, professor Anthony Sinskey, in his office. Credit: Grace van Deelen
When she first started her graduate work, neither Voigt nor Sinskey had expertise in enzyme biochemistry involving solid substrates: Sinkey’s lab focuses on bacterial metabolism, while Voigt’s lab focuses on genetic engineering (though Voigt did have experience with directed evolution research). Additionally, Zhong-Johnson’s path to her project was rather unconventional. Most grad students do not come to potential advisors proposing entire dissertations, which posed a unique challenge for Zhong-Johnson.
Despite not having specific expertise in enzyme biochemistry involving solid substrates, Voigt and Sinskey have supported Zhong-Johnson in other ways: by helping her to develop critical thinking skills and connecting her to other people in her field, such as potential collaborators, who can help her project thrive in the future. Zhong-Johnson has supplemented her MIT experience by having enzyme experts as part of her dissertation committee as well.
Sinskey says that, in the future — once Zhong-Johnson has engineered the ideal enzyme — they would like to partner with industry, and work on making the enzyme into a product that waste companies might use to recycle plastic. Additionally, Sinskey says, the plastic problem and the IsPETase solution raise so many interesting questions that Zhong-Johnson’s project will probably live on in the Voigt and Sinskey labs even after she graduates. He’d like to see other graduate students working to understand the enzyme’s activity and progressing the directed evolution that Zhong-Johnson started.
Zhong-Johnson is already working on understanding the specifics of how IsPETase act on PET. “How does it eat a hole in a plastic bottle? How does it move along and make the hole bigger as it moves through the process? Does it jump around? Or does it keep degrading a single polymer chain until its completely broken down? We just don’t know the answers yet,” says Sinskey.
But Zhong-Johnson is up to the task. “My graduate students have to have three skills, in my opinion,” Sinskey says. “One, they have to be intelligent. Two, they have to be energetic, and three, they have to be of high integrity, in research and behavior.” Zhong-Johnson, he says, has all three qualities.
Innovative brain-wide mapping study shows that “engrams,” the ensembles of neurons encoding a memory, are widely distributed, including among regions not previously realize
Picower Institute
April 12, 2022
A new study by scientists at The Picower Institute for Learning and Memory at MIT provides the most comprehensive and rigorous evidence yet that the mammalian brain stores a single memory across a widely distributed, functionally connected complex spanning many brain regions, rather than in just one or even a few places.
Memory pioneer Richard Semon had predicted such a “unified engram complex” more than a century ago, but achieving the new study’s affirmation of his hypothesis required the application of several technologies developed only recently. In the study, the team identified and ranked dozens of areas that were not previously known to be involved in memory and showed that memory recall becomes more behaviorally powerful when multiple memory-storing regions are reactivated, rather than just one.
“When talking about memory storage we all usually talk about the hippocampus or the cortex,” said co-lead and co-corresponding author Dheeraj Roy. He began the research while a graduate student in the RIKEN-MIT Laboratory for Neural Circuit Genetics at The Picower Institute led by senior author Susumu Tonegawa, Picower Professor in the Departments of Biology and Brain and Cognitive Sciences. “This study reflects the most comprehensive description of memory encoding cells, or memory ‘engrams,’ distributed across the brain, not just in the well-known memory regions. It basically provides the first rank-ordered list for high-probability engram regions. This list should lead to many future studies, which we are excited about, both in our labs and by other groups.”
In addition to Roy, who is now a McGovern Fellow in the Broad Institute of MIT and Harvard and the lab of MIT neuroscience Professor Guoping Feng, the study’s other lead authors are Young-Gyun Park, Minyoung Kim, Ying Zhang and Sachie Ogawa.
Mapping Memory
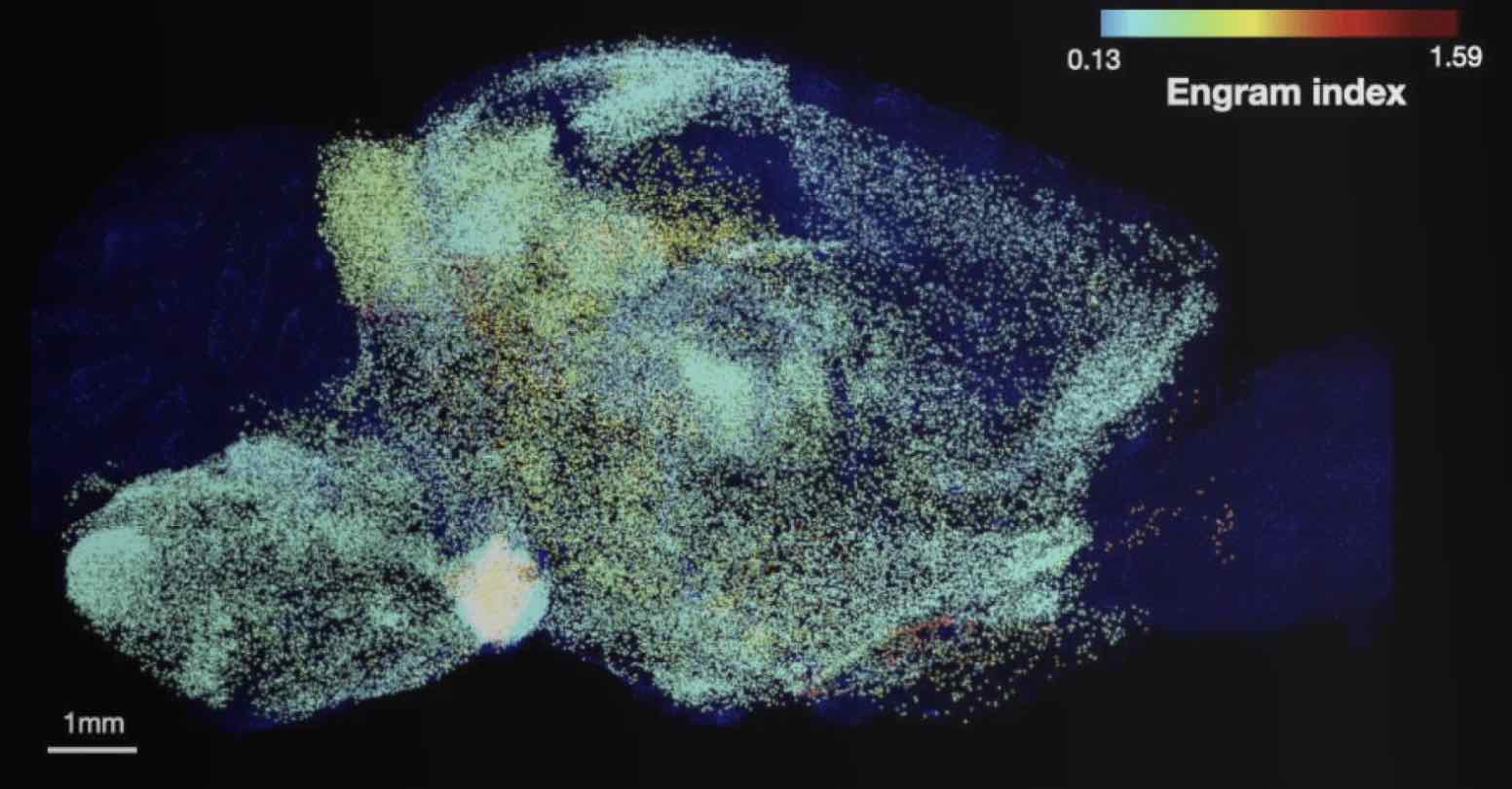
The team was able to map regions participating in an engram complex by conducting an unbiased analysis of more than 247 brain regions in mice who were taken from their home cage to another cage where they felt a small but memorable electrical zap. In one group of mice their neurons were engineered to become fluorescent when they expressed a gene required for memory encoding. In another group, cells activated by naturally recalling the zap memory (e.g. when the mice returned to the scene of the zap) were fluorescently labeled instead. Cells that were activated by memory encoding or by recall could therefore readily be seen under a microscope after the brains were preserved and optically cleared using a technology called SHIELD, developed by co-corresponding author Kwanghun Chung, Associate Professor in The Picower Institute, the Institute for Medical Engineering & Science and the Department of Chemical Engineering. By using a computer to count fluorescing cells in each sample, the team produced brain-wide maps of regions with apparently significant memory encoding or recall activity.
The maps highlighted many regions expected to participate in memory but also many that were not. To help factor out regions that might have been activated by activity unrelated to the zap memory, the team compared what they saw in zap-encoding or zap-recalling mice to what they saw in the brains of controls who were simply left in their home cage. This allowed them to calculate an “engram index” to rank order 117 brain regions with a significant likelihood of being involved in the memory engram complex. They deepened the analysis by engineering new mice in which neurons involved in both memory encoding and in recall could be doubly labeled, thereby revealing which cells exhibited overlap of those activities.
To really be an engram cell, the authors noted, a neuron should be activated both in encoding and recall.
“These experiments not only revealed significant engram reactivation in known hippocampal and amygdala regions, but also showed reactivation in many thalamic, cortical, midbrain and brainstem structures,” the authors wrote. “Importantly when we compared the brain regions identified by the engram index analysis with these reactivated regions, we observed that ~60 percent of the regions were consistent between analyses.”
Memory manipulations
Having ranked regions significantly likely to be involved in the engram complex, the team engaged in several manipulations to directly test their predictions and to determine how engram complex regions might work together.
For instance, they engineered mice such that cells activated by memory encoding would also become controllable with flashes of light (a technique called “optogenetics”). The researchers then applied light flashes to select brain regions from their engram index list to see if stimulating those would artificially reproduce the fear memory behavior of freezing in place, even when mice were placed in a “neutral” cage where the zap had not occurred.
“Strikingly, all these brain regions induced robust memory recall when they were optogenetically stimulated,” the researchers observed. Moreover, stimulating areas that their analysis suggested were insignificant to zap memory indeed produced no freezing behavior.
The team then demonstrated how different regions within an engram complex connect. They chose two well-known memory regions, CA1 of the hippocampus and the basolateral amygdala (BLA), and optogenetically activated engram cells there to induce memory recall behavior in a neutral cage. They found that stimulating those regions produced memory recall activity in specific “downstream” areas identified as being probable members of the engram complex. Meanwhile, optogenetically inhibiting natural zap memory recall in CA1 or the BLA (i.e. when mice were placed back in the cage where they experienced the zap) led to reduced activity in downstream engram complex areas compared to what they measured in mice with unhindered natural recall.
Further experiments showed that optogenetic reactivations of engram complex neurons followed similar patterns as those observed in natural memory recall. So having established that natural memory encoding and recall appears to occur across a wide engram complex, the team decided to test whether reactivating multiple regions would improve memory recall compared to reactivating just one. After all, prior experiments have shown that activating just one engram area does not produce recall as vividly as natural recall. This time the team used a chemical means to stimulate different engram complex regions and when they did, they found that indeed stimulating up to three involved regions simultaneously produced more robust freezing behavior than stimulating just one or two.
Meaning of distributed storage
Roy said that by storing a single memory across such a widespread complex the brain might be making memory more efficient and resilient.
“Different memory engrams may allow us to recreate memories more efficiently when we are trying to remember a previous event (and similarly for the initial encoding where different engrams may contribute different information from the original experience),” he said. “Secondly, in disease states, if a few regions are impaired, distributed memories would allow us to remember previous events and in some ways be more robust against regional damages.”
In the long term that second idea might suggest a clinical strategy for dealing with memory impairment: “If some memory impairments are because of hippocampal or cortical dysfunction, could we target understudied engram cells in other regions and could such a manipulation restore some memory functions?”
That’s just one of many new questions researchers can ask now that the study has revealed a listing of where to look for at least one kind of memory in the mammalian brain.
The paper’s other authors are Nicholas DiNapoli, Xinyi Gu, Jae Cho, Heejin Choi, Lee Kamentsky, Jared Martin, Olivia Mosto and Tomomi Aida.
Funding sources included the JPB Foundation, the RIKEN Center for Brain Science, the Howard Hughes Medical Institute, a Warren Alpert Distinguished Scholar Award, the National Institutes of Health, the Burroughs Wellcome Fund, the Searle Scholars Program, a Packard Award in Science and Engineering, a NARSAD Young Investigator Award, the McKnight Foundation Technology Award, the NCSOFT Cultural Foundation, and the Institute for Basic Science.
Greta Friar | Whitehead Institute
April 11, 2022
Cancer is at its most deadly when it spreads and forms tumors in new tissues. This process, called metastasis, is responsible for the vast majority of cancer deaths, and yet there is still a lot that researchers do not know about how and when it happens. Whitehead Institute Founding Member Robert Weinberg, also the Daniel K. Ludwig Professor for Cancer Research at the Massachusetts Institute of Technology, studies the mechanisms behind metastasis. One such mechanism is a process called the epithelial-mesenchymal transition (EMT), which causes epithelial cells, which normally stick tightly together, to lose their cohesion, enabling them to move around and even invade nearby tissue. This EMT program also operates during embryonic development. Cancer cells can co-opt this process and use it travel from their original tumor site to distant tissues throughout the body. Some of the cancer cells that spread are able, on rare occasions, to form new tumors in these tissues—metastases—while the great majority of these cells remain dormant after entering the distant tissues.
New research from Weinberg and postdoc Yun Zhang shows that cells change in diverse ways through the actions of the EMT, which can influence whether cells are able to form new tumors after they spread. The work, published in Nature Cell Biology on April 11, 2022, also identifies two regulators of the EMT and shows that loss of each regulator leads to a different metastatic risk profile.
“Using triple negative breast cancer as a model, we are trying to go a bit deeper into understanding the molecular mechanisms that regulate the EMT, how cells enter into different EMT intermediate states, and which of these states contribute to metastasis,” Zhang says.
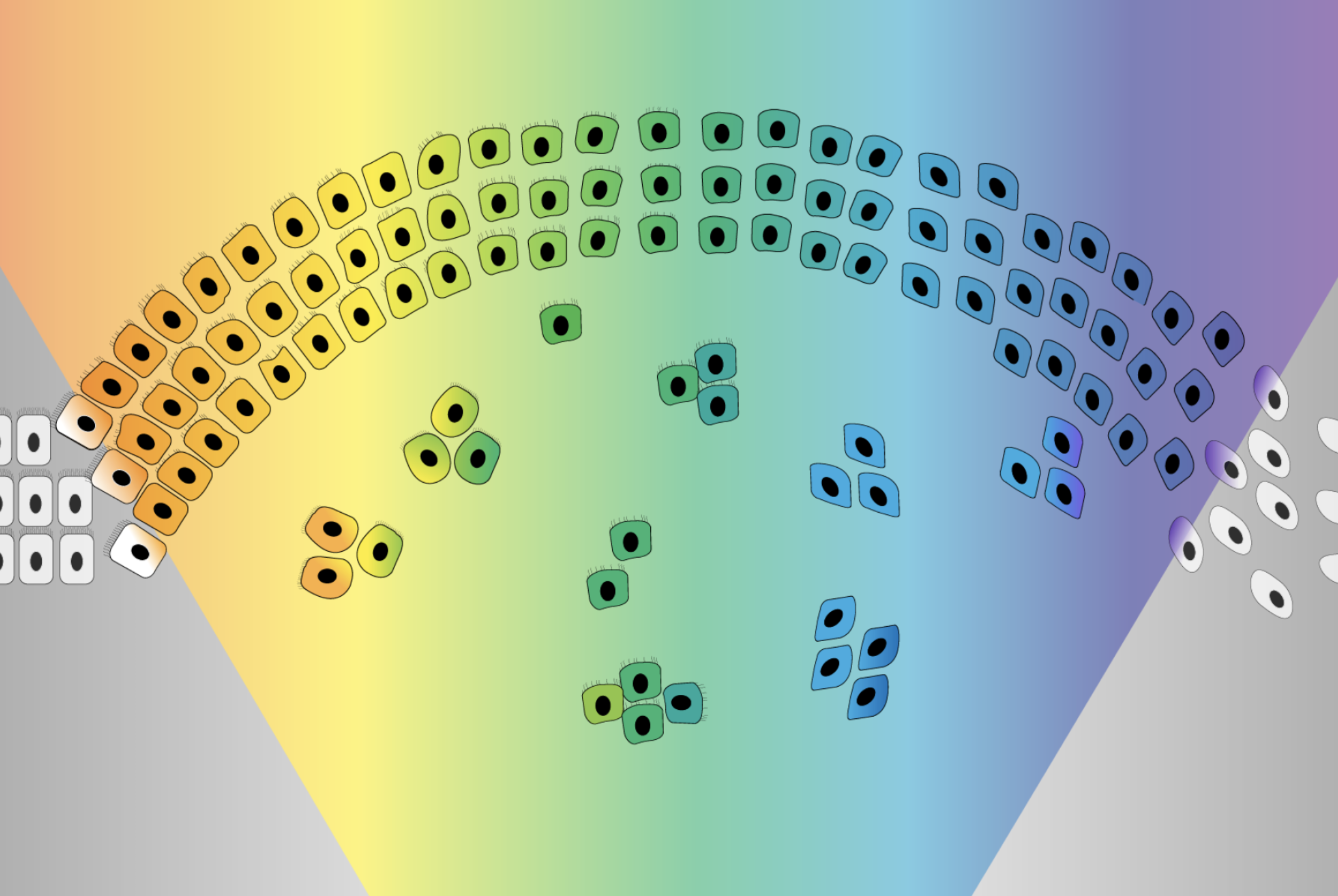
The EMT was originally imagined as a sort of binary switch, in which cells start out epithelial and become mesenchymal, much like a light switch being flicked from off to on. However, researchers are learning that the EMT works more like a dimmer switch that can be shifted along a spectrum of brightness. Cells that undergo the EMT usually end up in hybrid states between the epithelial and mesenchymal extremes. These cells in the middle of the spectrum, which have some characteristics of each extreme, are called “quasi-mesenchymal” cells, and it turns out that they–rather than cells that become fully mesenchymal–are the most capable of metastasizing and forming new tumors throughout the body.
Protected versus plastic cells
Weinberg and Zhang set out to better understand the EMT spectrum and what controls cells’ movement along it. First, they compared epithelial cells to each other and found that some were more plastic or prone to transitioning along the EMT spectrum than others. They also used the CRISPR gene editing tool to screen for genes that might be regulating the cells’ plasticity. If researchers can learn what makes a cell become quasi-mesenchymal—posing a high risk for metastasis—they might be able use this information, at some time in the future, to develop strategies to prevent cells from entering this high-risk state.
The CRISPR gene screen turned up a number of molecules that seemed to influence cells’ epithelial-mesenchymal plasticity. Two groups of these molecules had especially strong effects: PRC2, a complex that operates in chromosomes to silence or inactivate genes, and KMT2D-COMPASS, a complex that helps activate genes. Both complexes help to keep cells in a stable epithelial state. Loss of either complex makes cells more prone to moving along the EMT spectrum.
The researchers then determined how the loss of either complex enables the EMT. PRC2 normally silences several key EMT-related genes. When PRC2 is lost, those genes activate, which in turn sensitizes the cell to a signal that can trigger the EMT. The loss of KMT2D-COMPASS affects how well PRC2 can bind its targets, leading to the same signal sensitivity. In spite of the similar mechanisms at play, the loss of PRC2 versus KMT2D-COMPASS leads cells to transition to end up in different EMT states, an exciting finding for the researchers. Cells without KMT2D-COMPASS became fully mesenchymal, while cells without PRC2 became hybrid or quasi-mesenchymal. Consequently, cells without PRC2 were much more capable of metastasis than cells without KMT2D-COMPASS (or cells in which both complexes were active) in mouse models. When the researchers looked at historical data from breast cancer patients, they observed the same pattern: people with faulty PRC2 component genes had worse outcomes. These findings provide further evidence that cells in the middle of the EMT spectrum are most likely to metastasize.
This work supports the understanding of the EMT as a spectrum rather than a simple switch, and shows that different EMT regulators can program cells to transition to different parts of the EMT spectrum. Additionally, the finding that loss of PRC2 is linked to metastasis has implications for cancer drugs currently in development that work by inactivating PRC2. Benefits of the drugs may outweigh risks for patients with certain types of cancer for which PRC2 is an effective target. However, Weinberg and Zhang caution that researchers leading clinical trials of PRC2-targeting drugs should be careful about selecting patients and monitoring outcomes. In the types of cancer cells that the researchers looked at, even temporary PRC2 inactivation, such as from a therapy trial, was sufficient to trigger cells to become EMT hybrids with increased metastatic capacity.
Weinberg and Zhang intend to continue exploring the genes identified in their CRISPR screen to see if they can identify other hybrid states along the EMT spectrum, in which cells have different combinations of epithelial and mesenchymal features. They hope that by deepening their understanding of the gene expression profiles of cancer cells associated with different EMT trajectories, they can contribute to the development of therapies for people with potentially metastatic cancers.
“Understanding when and how cancer cells become able to form life-threatening metastases is crucial in order to help the many patients for whom this is a risk,” Weinberg says. “This work provides new insights into the mechanisms that enable cells to metastasize and the roles that different EMT programs can play.”
Depletion of either the DapB or Dxr proteins causes oxidative stress and cell death in bacteria, which could aid the development of more effective antibiotics.
Grace van Deelen
April 7, 2022
How do bacteria die? It’s an important question, especially since these single-celled organisms seem to be outpacing the development of new antibiotics. However, any one bacterial cell will often die from a number of separate but related pathways acting simultaneously, making understanding bacterial death difficult. Determining how to induce those pathways — and the role each pathway plays in a cell’s death — is key to creating effective antibiotics, especially after bacteria evolve to resist some drugs. But new research from Graham Walker’s lab in the MIT Department of Biology suggests that one historically under-appreciated cause of bacterial death, called oxidative stress, could help scientists develop antibiotics that kill bacteria more effectively.
Many antibiotics target the bacteria’s cell wall or replication process. However, some antibiotics can additionally cause changes in a cell’s metabolism that lead to a phenomenon called oxidative stress. Reactive oxygen-containing molecules float around freely inside the cell, sometimes bumping into other molecules, reacting with them, and stealing their unpaired electrons in a process called oxidation. For example, a guanine molecule — the DNA nucleotide commonly abbreviated to “G” — may become an oxidized guanine called 8-oxo-dG, a transformation that causes mutations in a cell’s genetic code. The harmful effects of oxidation are usually managed by the cell, but the disruption caused by these antibiotics can also become fatal to the cell.
In the case of 8-oxo-dG, the cell responds to this oxidative stress by attempting to cut the oxidized guanine out of the genome and repair it with a regular nucleotide during a process called base excision repair (BER). However, during BER, every completed step produces intermediate substances, including other forms of damaged DNA, which then must be cleared by another enzyme or protein. However, sometimes the cell is unable to complete BER because these intermediate substances build up. When there is an imbalance of intermediate substances, the cell pauses the repair, leaving breaks in the strands of DNA that cause cell death.
Because incomplete BER is just one of many contributing causes of cell death, the total contribution of incomplete BER to cell death remained unclear. As a result, scientists in the Walker lab were interested in determining other stressors, besides known antibiotics, that might cause incomplete BER of 8-oxo-dG. “If this mechanism of 8-oxo-dG getting into DNA causes bacteria to die, there’s probably some other stressor that isn’t an antibiotic that would cause cells to die by the same way,” says Walker.
In the paper, published on February 8 in mBio, the researchers determined two additional stressors that also induce cell death via incomplete BER of 8-oxo-dG. They found that the depletion of proteins DapB and Dxr also induced oxidative stress and incomplete BER of 8-oxo-dG. Scientists have known of these proteins — both of which are involved in bacterial metabolism — for some time, but had never associated them with incomplete BER.
“Incomplete base excision repair is probably one of more underappreciated ways a cell can die,” Walker says. “So we wanted to explore that pathway further.”
Charley Gruber, a postdoc in the Walker lab and lead author on the paper, identified DapB and Dxr by screening a library of 238 proteins essential for Escherichia coli growth. He determined that, in the absence of these two proteins, the cell overproduced the reactive oxygen-containing molecules that contribute to oxidative stress. As a result, the oxidized nucleotide 8-oxo-dG was incorporated into the genome, leading to cell death through incomplete BER. Researchers don’t know for sure why depletion of DapB and Dxr increases the amount of reactive oxygen-containing molecules inside the cell, but oxidative stress is a common reaction to many disruptions that bacterial cells may face.
To Walker and Gruber’s surprise, their results also showed that the total contribution of incomplete BER to cell death was different between the two proteins — Dxr-depleted cells died faster than DapB-depleted cells, suggesting that a lack of Dxr played a larger role in cell death. Because the responses to protein depletion were so different between DapB and Dxr, the researchers concluded that there is no singular pathway that causes oxidative stress; rather, it is probably a common consequence of many possible disruptions to bacterial cell physiology.
“If there’s one important thing I think we need to realize about cell death,” Gruber says, “it’s that a lot is happening to a stressed cell. And what is actually lethal might differ between two cells.”
This study adds to a body of research by Gruber, Walker, and others about the role of incomplete BER in the process of cell death. In 2012, the Walker lab published a paper in Science — building on earlier work from MIT’s Termeer Professor of Bioengineering, Jim Collins — which showed for the first time that some commonly-used antibiotics kill by way of oxidative stress and the 8-oxo-dG pathway of incomplete BER. The idea was not immediately accepted by the scientific community, and a debate ensued: Shortly after Walker’s paper, Northeastern University biologist Kim Lewis and University of Illinois biologist Jim Imlay each published separatepapers suggesting that bactericidal antibiotics had nothing to do with oxidative stress. Since then, the Walker and Collins labs have continued to research the topic, producing more supporting data for their argument that oxidative stress and incomplete BER are, in fact, an important pathway of cell death.
“This new work provides a strong genetic foundation for the role of incomplete BER in bacterial cell death,” Collins says . “Oxidative stress and BER should be targeted as a means to potentiate existing antibiotics and enhance our antibiotic arsenal.”
Scientific debates like the one surrounding the contribution of incomplete BER to bacterial death are crucial to the creation of effective antibiotics. Most antibiotics work by breaking the cell wall and causing cell death that way. However, the lab’s findings offer a possibility for antibiotic assistance: the common practice of using secondary antibiotics to aid in cell death thorough a different pathway. For example, administering a secondary antibiotic that triggers the 8-oxo-dG pathway along with the primary antibiotic that is lethal to bacteria through cell wall destruction could be more effective than one antibiotic on its own, Gruber suggests.
“Many of our antibiotics are not working, or we’ve overused them in some cases, so we’re really running out of drugs,” he says. “So an antibiotic that induces oxidative stress could be another way to help existing drugs work better.”
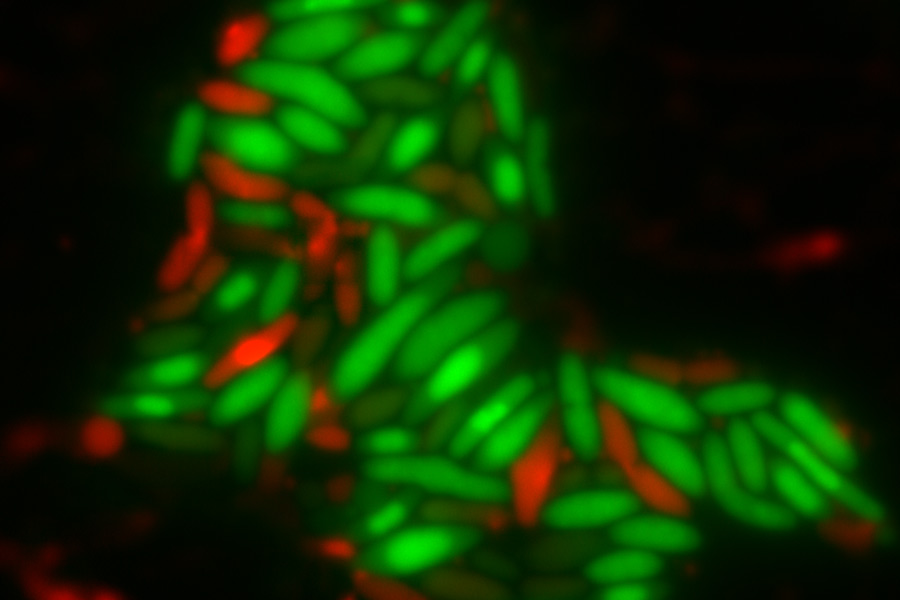
Top image: E. coli cells with either DapB (left) or Dxr (right) depleted. Living cells are stained green while dead cells are stained red. Credit: Charley Gruber
Citation: “Degradation of the Escherichia coli Essential Proteins DapB and Dxr Results in Oxidative Stress, which Contributes to Lethality through Incomplete Base Excision Repair” mBio, online February 8, 2022, DOI: 10.1128/mbio.03756-21
Charley C. Gruber, Vignesh M. P. Babu, Kamren Livingston, Heer Joisher, and Graham C. Walker
Institute Professor Sallie “Penny” Chisholm is best known for her role in discovering the tiny bacteria called Prochlorococcus — the world’s most abundant photosynthetic organism. But she has also played a pivotal role in pioneering and advocating for women’s rights at MIT and beyond.
Celina Zhao
March 31, 2022
Without the ancestors of the ocean microbe that Sallie “Penny” Watson Chisholm discovered in 1986, humans may not have ever evolved on Earth. The tiny microbe is called Prochlorococcus, and it’s full of superlatives. One hundred of them can fit on the width of a single strand of human hair, making them the smallest photosynthetic organisms on the planet. At the same time, they’re also the most abundant, comprising an integral piece of the oceans’ “invisible forest.” In fact, you can thank them for the oxygen you breathe in every twentieth breath you take.
With their population, which numbers in the billion billion billions and weighs a collective 220 million Volkswagen Beetles, you might expect Prochlorococcus to be easy to find. But they were not uncovered until the 1980s — and by accident, at that. Since that fateful discovery, Chisholm has dedicated her life and career to studying these intriguing little cells. They have earned her a National Medal of Science from former President Barack Obama, led her to a debate with the Dalai Lama, and even sparked a meeting with the Wu-Tang Clan’s GZA, a rapper interested in featuring the bacterium in his album.
This extraordinary journey, however, was not a straight path — in part because being a pioneering female researcher was no easy feat.
Early life and education
Chisholm was born on November 5, 1947 in Marquette, Michigan, a small town located on the shores of Lake Superior. While growing up, her passion was not science, but skiing, and when it came time to apply for college, she had little ambition and few dreams in higher education.
Her parents, however, intervened. Chisholm’s mother, a traditional 1950s housewife, hated not having her own job or income. In particular, she stressed to her daughter the importance of being able to get an education and career. So, Chisholm traveled east to Saratoga Springs, New York, enrolling at Skidmore College — then a private liberal arts women’s college.
Chisholm on the RV Ellen B. Scripps during her postdoctoral years at the Scripps Institute of Oceanography. Credit: David Karl, University of Hawaii
Living at Skidmore, where she studied biology and chemistry, was a formative experience. “It unconsciously builds a confidence,” Chisholm says of the absence of men competing for attention and resources. In her senior year, she participated in her first fieldwork expedition, studying the prevalence of the mineral manganese in a local lake. Noticing her interest and potential in research, her advisor suggested she attend graduate school. Though the idea had never occurred to her before, it sounded much more interesting than entering the workforce, and so she agreed.
After a year as a graduate student at Cornell University, Chisholm transferred to the State University of New York (SUNY) to study freshwater plankton. With her PhD in hand — the first PhD in her entire extended family — she moved to the sunny beaches of La Jolla, California for her postdoctoral studies. At the University of California San Diego’s Scripps Institute of Oceanography, she began studying the massive system to which she’d dedicate the rest of her career: the ocean.
Just a few months after arriving at Scripps, Chisholm sailed around the Gulf of California on her first research cruise, the R/V Alpha Helix. She stood out from most of the members of the ship in a prominent way — she was one of only a few women aboard. Women were only just starting to be allowed on research vessels in the 1970s, an imbalance that would persist on Chisholm’s other cruises at Scripps.
And, though she loved her time at Scripps, by 1976 it was time to move on. She had three options: a small marine lab in Maine, an oceanography department in Canada, or the Civil and Environmental Engineering Department at MIT. In the case of the latter, she would not simply be the only biologist; she would be the only woman. Should she choose a comfortable route, or should she choose the challenge? Then, one of her mentors at Scripps told her, “Penny, you don’t turn down MIT.” It was a once-in-a-lifetime opportunity, so she decided to give it a shot.
Settling into MIT
In 1976, Chisholm arrived at MIT to continue studying the physiology of various phytoplankton species. One of the instruments she used was a flow cytometer. Though traditionally only used in medical settings, she’d discovered that flow cytometers — with their ability to move individual cells in single file past a laser for a convenient close-up view — were also excellent for her research. In addition, she’d noticed that the unique pigments of phytoplankton fluoresced distinctive colors in the laser’s presence. For example, the green chlorophyll found in phytoplankton would emit red light when struck by a blue laser. Accessory pigments in some would emit orange light.
One day, she and her team came to a game-changing idea: “Wouldn’t it be cool if we could take an instrument like this out on a ship and just squirt sea water through it to see what the diversity of phytoplankton look like?” she mused. This idea immediately opened a whole new set of doors for possible research.
At the start of Chisholm’s career, scientists had a highly restricted picture of microbes in the ocean. This was largely due to limitations in microscope technology: phytoplankton smaller than 5 microns were extremely difficult to see using standard microscopy of the day. In 1979, however, John Waterbury of the Woods Hole Oceanographic Institution, using a technique known as epifluorescence microscopy, discovered tiny photosynthetic cyanobacteria only about 2 microns in diameter, which glowed orange. He named the bacterium Synechococcus. Chisholm was intrigued, and her team set off to the Caribbean in 1985 with their flow cytometer to study them.
While studying images of Synechococcus, they started seeing extremely tiny red signals on their instrument. They didn’t think much of it at first — assuming it was probably just electronic noise. But, as these signals kept showing up, they began to wonder: Could these signals be coming from something that was alive?
The Department of Civil and Environmental Engineering faculty picture from 1978 shows that Chisholm (middle, second row from the back) was the only female faculty member.
It was an intriguing possibility, with evidence in its favor. She and her team noticed that the signals varied depending on the depth and temperature of the water sample being analyzed. There was no reason for electronic noise to increase with depth. But there was good reason for cells to have more chlorophyll (hence the red color under the blue laser of the flow cytometer) the deeper they were under the surface of the water, in order to help with photosynthesis.
Over the next few years, collaborators discovered the same little cells in other seas across the globe. In 1988, Chisholm and her team published their findings in the journal Nature. In the paper, the researchers referred to them as a “new group of pico-plankters,” but members of the lab affectionally called them “little greens” because they contained chlorophyll b, which is a characteristic of green plant chloroplasts. For its formal name, they eventually chose “Prochlorococcus,” meaning “little round progenitors of chloroplasts,” or more colloquially “primitive green berries.”
Chisholm ultimately refocused her entire lab on Prochlorococcus, which revealed itself to be a fascinating subject. She had been looking for a phytoplankton species that could be both easily studied in the lab and easily found in the oceans. Prochlorococcus seemed like a promising model system through which she and her lab could begin understanding how the ocean works.
It turns out that Prochlorococcus wasn’t just one uniform organism, but a collective, composed of more than 30 clusters of strains called “ecotypes” with their own unique survival tactics. This collective has adapted to various environments, dividing up vast swaths of the oceans with various light, temperature, and nutrient combinations. Each ecotype is the “most efficient photosynthetic machine” at its particular conditions — and added together produce 10% of the oxygen in the atmosphere.
To Chisholm, this was an unmistakable sign that there was something truly remarkable about Prochlorococcus. But it would take a few more years for technology to develop far enough to uncover more.
The intriguing new cells glowed red under the flow cytometer (left), but appeared like mere specks of dust or electronic noise under the commonly-used light microscopes (right). Credit: Rob Olson, Woods Hole Oceanographic Institute
Taking a stance on women’s rights at MIT
Deep in the thralls of Procholorococcus research, Chisholm worked early mornings and stayed late into the night. She was in her late forties, and her life revolved around her job. In 1994, during one of those typical long days in her office, she received a phone call that dramatically changed the way she viewed her career.
It was from Nancy Hopkins, a fellow MIT professor whom she casually knew. Hopkins was rallying the support of the 17 other senior female faculty at MIT for a purpose: She believed MIT was discriminating against all of them in multiple areas, including lab space, pay, and support. Hopkins asked Chisholm if she would be willing to come to a meeting and discuss these issues.
Chisholm didn’t know what would come of the meeting, nor did she expect much change to happen. In fact, she had never really thought about what it meant for her to be among the few female faculty at MIT. “I was just doing my work,” she says, “and I liked being one of the guys. I wasn’t plugged into feminist issues or anything.”
But she agreed to attend, and in a few days, found herself in a discreet location on campus. The room was small. Some women sat on chairs, while others sprawled on the floor.
It was awkward at first. “We didn’t know each other that well, so nobody wanted to speak out. We didn’t want to be that one woman faculty member to complain because that meant you couldn’t cut it,” she recalls. But soon everyone started shedding their protective shells. Hours went by as they all found common ground.
Chisholm came to a realization, which she later articulated in her Killian Award acceptance speech: “There’s always a general sense of not being part of the club. It’s like there’s a playbook for this whole enterprise we’re involved in, and it was written by men. As a woman, you’re just constantly trying to figure out what the game is and what the playbook is.”
In her office in 1988, Chisholm holds an image of Prochlorococcus. Credit: Donna Coveney
Armed with a letter calling for investigation, they marched into Dean Robert J. Birgeneau’s office. One of the women even put on a skirt for the first time. They had no idea how he’d react, but preliminary evidence seemed to be on their side. Not only were there 194 tenured male professors compared to only 15 tenured women faculty in the School of Science, the percentage of female faculty hadn’t budged from 8% in 20 years.
Seeing their determination and the evidence, Birgeneau agreed to help. Together with MIT President Charles Vest, he established the MIT Committee on Women Faculty in the MIT School of Science. Birgeneau and Vest charged the committee “to study the status of women in science at MIT and, among other things, to determine the reasons for [MIT’s] failure in the School of Science to hire and promote significant numbers of women faculty.”
The committee’s findings over a two year period were published in a report in 1999. The report made headlines in nearly every major newspaper when President Vest admitted to and apologized for the gender discrimination.
“It was a shot heard around the world,” Chisholm says. And it was true — the MIT administration acted quickly and decisively to right its wrongs. Salaries were adjusted, space and equipment allocation were corrected, retirement packages were increased, among many other changes. Importantly, several other higher education institutions followed MIT’s lead.
As a result of their shared experience, the group of women faculty grew close. Chisholm says, “In my mind, that was one of the best outcomes — on top of all the positive changes by the administration — in terms of my quality of life at MIT: having a community of women that I could talk to.”
Increasing recognition for Prochlorococcus and legacy at MIT
Parallel to the push for equity for women faculty at MIT was the development of one of the most ambitious science expeditions to date — sequencing the human genome. After the Human Genome Project released the sequence of all 20,000 human genes in 2003, there was an abundance of DNA sequencing machines available for other projects. Chisholm leapt at the chance to learn more about her “little greens.” A strain of Prochlorococcus called MED4 became the second microbial genome to be sequenced.
The sequencing “completely opened up the black box,” Chisholm says. With only 1,700 genes, that strain of cyanobacteria is one of the simplest self-sustaining organisms known. Chisholm wrote in a chapter of “Microbes and Evolution: The World that Darwin Never Saw”, “This cell is truly the ‘essence’ of life. As a photosynthesizer, it can do what humans cannot, even with all of our technology: It can split water using sunlight and make hydrogen and oxygen — all with only 1,700 genes.”
The cyanobacterium Prochlorococcus was Chisholm’s muse throughout the majority of her career. Credit: Luke Thompson and Nicki Watson
Together, Chisholm and her lab eventually learned that though each Prochlorococcus strain contains fewer than 2,000 genes, the collective as a whole contains more than 80,000 — four times the size of the human genome. There is a core set of genes (about 1,200) that all Prochlorococcus share, and a few hundred more that are shared only by a subset of strains. In addition, each individual strain has an additional 80-200 genes that are completely unique to it. That incredible diversity allows Prochlorococcus to thrive in all sorts of environmental conditions, from 40 degrees north to 40 degrees south latitudes to almost 200 meters under the ocean where there’s less than 1% light penetration. As the environment shifts, so does the ecotype composition, stabilizing the total population of the species.
Additionally, it turns out that Prochlorococcus is also integral to its local environment. The 5 billion tons of living biomass it produces though photosynthesis each year is eaten by small microorganisms, then zooplankton, then fish. Ultimately, Prochlorococcus feeds 10% of all the creatures in the sea.
Over the years, Chisholm has led the charge in uncovering more about these intriguing cyanobacteria, including how they interact with other things in their environment like viruses or different microorganisms, as well as the unusual molecules Prochlorococcus produce. This persistent dedication has earned her many awards and honors. In 2011, former President Barack Obama presented her with the National Medal of Science, the White House’s highest honor for American scientists. In 2014, she won the MIT James R. Killian Faculty Award, and in 2015 she was named an MIT Institute Professor — the highest title that MIT bestows, and one that only 13 faculty presently hold. In 2019, she was awarded the Crafoord Prize in Biological Sciences by the Swedish Academy of Sciences, the equivalent of a Nobel Prize for biosciences.
As she was gaining worldwide recognition for her research, Chisholm also served as teacher and mentor to many classes of students at MIT. In 1993, she joined the Department of Biology to teach 7.014 (Introductory Biology). Her class was the only introductory biology class to feature ecology. In her lab, she advises undergraduate and graduate students with varying backgrounds, from biology, to chemistry, to oceanography, to civil and environmental engineering.
Her advocacy for women faculty’s rights at MIT has also paid off over the years, with statistics slowly but surely improving to more equal footing. As of June 2019, 250 of the approximately 1,050 faculty members are women, and within the Schools of Science and Engineering, women comprise 22% of the faculty.
And, nearly four decades later, Prochlorococcus still remains an irresistible siren to her. “I should probably be thinking about retiring, but I’m not because Prochlorococcus is too darn interesting,” Chisholm says. “I’m really very grateful to have this organism in my life.”
Posted: 3.31.22
March 29, 2022
Emily Makowski | Ragon Institue
March 25, 2022
Dr. Alison Ringel didn’t set out to exclusively choose female mentors when she started out in scientific research, but by following her research interests, that’s what happened. “I have been mentored by what I call a dream team of women faculty,” she says.
Ringel joined the Ragon in January 2022 as one of three faculty recently hired in newly created joint appointment positions between the Ragon Institute and the Department of Biology at Massachusetts Institute of Technology. She currently studies how T cells — white blood cells that help protect against infection and can help fight cancer — survive in stressful environments. Originally from Wilton, CT, Ringel’s first research experience was in yeast genetics as an undergrad at Wesleyan University. “I was really given a lot of freedom to pursue my own ideas … having that very early exposure and freedom to manipulate systems experimentally, make hypotheses, make mistakes, really solidified my interest,” she says.
Ringel graduated with two bachelor’s degrees — one in molecular biology and biochemistry, and one in physics — and completed a molecular biophysics PhD at Johns Hopkins University School of Medicine in the lab of structural biologist Cynthia Wolberger. She studied how chromatin-modifying enzymes — enzymes that modify the material that our chromosomes are made of, affecting gene expression — choose which proteins to modify. The experience was a very positive one. “Not only was that an incredibly rich environment, but she was also a very supportive mentor, and still is a supportive person in my life,” Ringel says.
As Ringel was finishing up her PhD, she became more interested in the small molecule cofactors that catalyze reactions involving these enzymes. She started to look into labs studying immunology and metabolism, and she met cell biologist Marcia Haigis, who became Ringel’s postdoctoral mentor at Harvard Medical School (HMS). Ringel, Haigis and their collaborators identified a single gene change in cancerous tumor cells that prevented T cells from functioning properly in obese mice, suggesting that obesity can have an impact on the immune system’s ability to recognize and clear a tumor.
Ringel also became a close collaborator with Arlene Sharpe, chair of the Immunology Department at HMS. Seeing how both Haigis and Sharpe were able to balance success in science with fulfilling personal lives has been a great inspiration to Ringel, who has two young sons. “Working for women who had done the whole thing — they had been amazing scientists, they had made significant discoveries that advanced their field, they all had families, they all had navigated tenure processes at really big, impactful research institutions — has been enormously helpful and inspiring to me throughout my career trajectory,” she says. Her advice for women in STEM and STEM-related fields? “Don’t be dissuaded by the challenges. Understand them and use them for your forward momentum.”
Ringel has been enjoying starting her own lab in her dual-appointment role and is currently looking at how T cells can restrict the growth of tumors. “I’m particularly excited about this connection between MIT Biology and the Ragon Institute,” she says. “I was so excited to land here because the Ragon Institute is a real powerhouse in everything immunology, and MIT Bio has the immense depth and breadth of scientific interest. This position was really unique in having both of those arms represented.”
Whitehead Institute
March 25, 2022
For the hundreds of thousands of people diagnosed with breast cancer each year, surgery to remove the cancerous tissue is often the best option — but this relatively simple procedure comes with some drawbacks. In more than a few cases, the surgical removal of a tumor can lead to an increased risk of the cancer reemerging in other locations in the body.
In a 2018 study, a postdoc in the lab of Whitehead Institute Member Bob Weinberg discovered that, at least in mice, this phenomenon was due to a bodily butterfly effect: the creation of a wound site in one place in the body, which necessitated subsequent wound healing, caused immune system changes affecting distant parts of the body.
These changes occurred as bone marrow cells responded to the wounding with a flood of inflammatory cells that entered into the wound site and, at the same time, scattered throughout the body. These dispersed inflammatory cells weakened the ability of the immune system to control the outgrowth of a distantly located metastatic tumor. Without this immune control, which otherwise could keep the metastasis at a very small size, the metastasis would grow out aggressively.
Hence, wounding in one part of the body provoked metastasis outgrowth at a distant site. This suggested, among other things, that the outgrowth of metastatic tumors, which is often seen in women who have recently undergone a mastectomy, might be actively provoked by the post-surgical wound-healing process.
Weinberg’s work also presented a way to potentially avoid this effect, using a preventative measure that’s probably sitting in your bathroom cabinet right now: the cheap and common class of drugs known as NSAIDS, which includes ibuprofen and aspirin. When mice were given NSAIDS before and after tumor removal surgeries, they experienced a fivefold lower rate of cancer recurrence at the site of metastasis than a control group given opioids. These NSAIDs could therefore be used in place of the opioids, which are often used to treat post-surgical pain.
The human body is full of undiscovered connections like this one and adding in foreign substances further complicates matters. While a treatment might work well in a Petri dish, researchers describe whole -body metabolism as “a whole different kettle of fish.”
The way drugs move through the body and interact with internal systems is called pharmacokinetics. When a person is given a medicine — either orally, through a chemotherapy method, or via injection — that drug must be able to find its way to its target in a high enough concentration to have an effect, and then when its purpose is served, it must be able to leave the body safely and not build up to a harmful amount.
Much like Weinberg’s work on NSAIDS in breast cancer, Whitehead Institute’s basic research has led to other surprising discoveries about drug activities in the human body. Read on to learn about research that is changing the way new drugs are designed, making existing treatments less toxic, and more.
Concentration is key
When it comes to the action of drugs in the human body, concentration is key. Just ask Rick Young, a Whitehead Institute Member and professor of biology at MIT. In 2018, Young’s lab, which had previously studied the regulatory circuitry involved in transcription (the copying of DNA into RNA), shifted its focus after discovering tiny droplets within cells that concentrate the molecular materials needed to transcribe the DNA.
The droplets, called transcriptional condensates, were the newest in a slew of recent discoveries of other such groupings of cellular components. Some of these aggregations facilitate RNA splicing while others help to form ribosomes.
For Young, the discovery of transcription-related condensates sparked an interest in how these droplets were affecting the action of drugs. Previous theories held that transcription was able to take place in cells because there was a sufficient concentration of necessary proteins, such as RNA polymerase and other accessory proteins. As the Young lab showed, these collaborating cellular players were actually being concentrated in the condensates,
In 2020, Young and Ann Boija and Isaac Klein, two postdocs in his lab, took their investigation a step further, analyzing the mechanism by which several cancer drugs are concentrated in cellular condensates, and how that concentration could affect their action in individual cells and thus in the body. They found that cancer drugs sort themselves into specific types of condensates, independently of their targets, which can allow them to build up into high concentrations in these localized areas within cells.
“This could have enormous implications for the way we discover and develop drugs,” said Rick Young. “If drugs had properties that had them partitioning into a condensate where their target lives, then they would enjoy two properties of condensates: they would be compartmentalized, and they would be at much higher concentrations than if they diffuse through the cell.”
Young’s work on condensates led him to co-create a pharmaceutical company called Dewpoint Therapeutics, with the goal of reformulating treatments for cancer or neurological conditions such as amyotrophic lateral sclerosis by targeting biomolecular condensates. Whitehead Institute Founding Member Rudolf Jaenisch serves as a scientific advisor.
Trouble in parasites
While researchers in Young’s lab investigate how drugs could be more efficiently targeted, Sebastian Lourido’s lab is taking a different tack — why do some drugs stop working as time progresses?
The malaria drug artemisinin was developed in China in the 1970s, and completely changed the way the world treated malaria. In the following decades, however, the parasites that cause malaria, several species within the genus Plasmodium, have slowly grown less susceptible to the drug.
In a paper published in September of 2020, Whitehead Institute Member Lourido and collaborators identified two parasite genes that were negatively impacting the actions of the drug in the parasite’s cells.
Researchers liken artemisinin to a “ticking time bomb,” which needs another molecule, called heme, to light its fuse. Heme, a small molecule that is one component of hemoglobin, helps transport electrons and deliver oxygen to tissues. When heme encounters artemisinin, it activates the drug, allowing the creation of small, toxic chemical radicals. These proceed to react with the parasites proteins, fats, and metabolites, eventually leading to its death.
In order to understand how some parasites were becoming less vulnerable to the drug, Lourido, along with researchers Clare Harding, Boryana Petrova and Saima Sidik, ran a genetic screen on a related parasite, Toxoplasma gondii. The screen allowed them to assess which mutations in the parasites’ genomes were beneficial for their survival and which ones were harmful.
The screen revealed two genes that affected how susceptible the parasites were to treatment with artemisinin. One, called Tmem14c, seemed to be protecting the parasites. The gene is analogous to a gene that transports heme out of mitochondria where it is generated. Lourido hypothesized that when the Tmem14c protein is working properly, it helps the cells shuttle heme and its building blocks and get them where they need to go in the cell. When this gene is knocked out or mutated, heme can build up in the parasite cells, making them more likely to activate the artemisinin “bomb.”
Another gene, when mutated, made the parasites less sensitive to artemisinin. The gene, called DegP2, encodes a protein that plays a role in heme metabolism, so when it was mutated, less heme was available in the cells to activate the drug.
This knowledge provides useful insights for treatment methods, said Lourido. For example, healthcare providers should take into consideration the fact that heme is key in artemisinin’s action, and avoid combining the drug with other treatments that might lower the amount of heme in parasite cells. “Understanding how different pathways within the cell participate to render parasites susceptible to these antiparasitic drugs helps us better pair them with other compounds that are going to be synergistic and not work against our own goal of defeating parasites,” Lourido said.
Taking the edge off toxic treatments
Another application of fundamental pharmacokinetics research involves mitigating the harmful effects of drugs. Consider the chemotherapy drug methotrexate. Methotrexate was the very first targeted drug ever made. Developed more than 60 years ago by Dr. Sidney Farber, the drug acts by inhibiting a key molecule in the metabolic process that builds DNA and RNA, thereby interfering with basic functions of the cell and with DNA synthesis, repair and replication, helping to destroy cancerous cells in the body.
Methotrexate is still a widely used component of chemotherapy cocktails, especially for pediatric leukemia. In the human body, though, methotrexate is like a bull in a china shop. It is very effective at knocking back cancer, but the drug’s life-threatening side effects, including gut, liver, kidney and brain damage, often lead doctors to terminate their patients’ treatment early, or seriously compromise the survivors’ quality of life.
The drug was much-studied in the 70s, but research trailed off in the subsequent decades due to limits on the existing technologies. Nearly fifty years later, Naama Kanarek, then a postdoctoral researcher in the lab of former Whitehead Institute Member David Sabatini, decided to take a fundamental research approach to studying the effects of methotrexate, in hopes that she might discover some way to make the drug less toxic.
“We now have access to genetic tools that allow us to address long standing questions in a way that was not possible before,” said Kanarek, who now runs her own lab at Boston Children’s Hospital. “We can use a CRISPR screen, and instead of focusing on what is known, we can ask what is unknown about the drug. We can find new genes that are involved in the response of cells to the drug that were not found before simply because the tools were not there.”
The screen revealed one gene in particular that seemed to be playing a role in how sensitive cancer cells were to methotrexate, the researchers reported in Nature in July of 2018. The cells’ sensitivity is important, Kanarek said, because if the cells can be made more vulnerable to methotrexate, the duration of treatment or required dose could be reduced. “If we can reduce dose because we can improve efficacy, then we can reduce toxicity without compromising on the cure rates and that is good news to the patients,” Kanarek said.
The gene in question, called FTCD, encodes an enzyme involved in the breakdown of the amino acid histidine. When the gene was knocked out, cancer cells were less sensitive to methotrexate. When the pathway was boosted with the addition of extra histidine, cells became more sensitive.
Former Whitehead Institute Director Susan Lindquist, who passed away 2016, performed similar work on the natural product amphotericin B, a drug which is used to treat some fungal infections. The drug is especially useful because fungi have not yet developed a resistance to it, as they have with so many other treatments. But amphotericin B also has some serious drawbacks; it can cause kidney damage, heart failure, and other serious and even fatal side effects.
These side effects mostly happened because amphotericin B works by binding to a chemical group called a sterol. In fungi, it binds to molecules called ergosterols in the cell wall, destabilizing the cells. Unfortunately humans also have a prevalent sterol: cholesterol. When amphotericin B binds to cholesterol in human cell membranes, it can damage human cells.
Using chemical synthesis methods, Lindquist and colleagues at Whitehead Institute and elsewhere were able to tweak the structure of the drug to bind only to ergosterol molecules and not cholesterol, bypassing most of the harmful side effects.
Why fundamental research
Drug development is often an extremely targeted pursuit, but for Whitehead Institute scientists, their advances have mostly come from a simple curiosity about the cellular mechanisms. For example, Rick Young didn’t set out to study condensates, but an inquiry into the fundamentals of transcription led him in this entirely new direction.
Such fundamental research has the potential to branch in any number of different ways. “Fundamental science can lead in directions that you would not foresee,” said the Institute’s Associate Director of Intellectual Property Shoji Takahashi. Basic research into drug behavior is essential and can contribute to life-changing therapies down the line.