New discovery suggests that all life may share a common design principle.
Justin Chen | Department of Biology
March 29, 2018
By studying bacteria and yeast, researchers at MIT have discovered that vastly different types of cells still share fundamental similarities, conserved across species and refined over time. More specifically, these cells contain the same proportion of specialized proteins, known as enzymes, which coordinate chemical reactions within the cell.
To grow and divide, cells rely on a unique mixture of enzymes that perform millions of chemical reactions per second. Many enzymes, working in relay, perform a linked series of chemical reactions called a “pathway,” where the products of one chemical reaction are the starting materials for the next. By making many incremental changes to molecules, enzymes in a pathway perform vital functions such as turning nutrients into energy or duplicating DNA.
For decades, scientists wondered whether the relative amounts of enzymes in a pathway were tightly controlled in order to better coordinate their chemical reactions. Now, researchers have demonstrated that cells not only produce precise amounts of enzymes, but that evolutionary pressure selects for a preferred ratio of enzymes. In this way, enzymes behave like ingredients of a cake that must be combined in the correct proportions and all life may share the same enzyme recipe.
“We still don’t know why this combination of enzymes is ideal,” says Gene-Wei Li, assistant professor of biology at MIT, “but this question opens up an entirely new field of biology that we’re calling systems level optimization of pathways. In this discipline, researchers would study how different enzymes and pathways behave within the complex environment of the cell.”
Li is the senior author of the study, which appears online in the journal Cell on March 29, and in print on April 19. The paper’s lead author, Jean-Benoît Lalanne, is a graduate student in the MIT Department of Physics.
An unexpected observation
For more than 100 years, biologists have studied enzymes by watching them catalyze chemical reactions in test tubes, and — more recently — using X-rays to observe their molecular structure.
And yet, despite years of work describing individual proteins in great detail, scientists still don’t understand many of the basic properties of enzymes within the cell. For example, it is not yet possible to predict the optimal amount of enzyme a cell should make to maximize its chance of survival.
The calculation is tricky because the answer depends not only on the specific function of the enzyme, but also how its actions may have a ripple effect on other chemical reactions and enzymes within the cell.
“Even if we know exactly what an enzyme does,” Li says, “we still don’t have a sense for how much of that protein the cell will make. Thinking about biochemical pathways is even more complicated. If we gave biochemists three enzymes in a pathway that, for example, break down sugar into energy, they would probably not know how to mix the proteins at the proper ratios to optimize the reaction.”
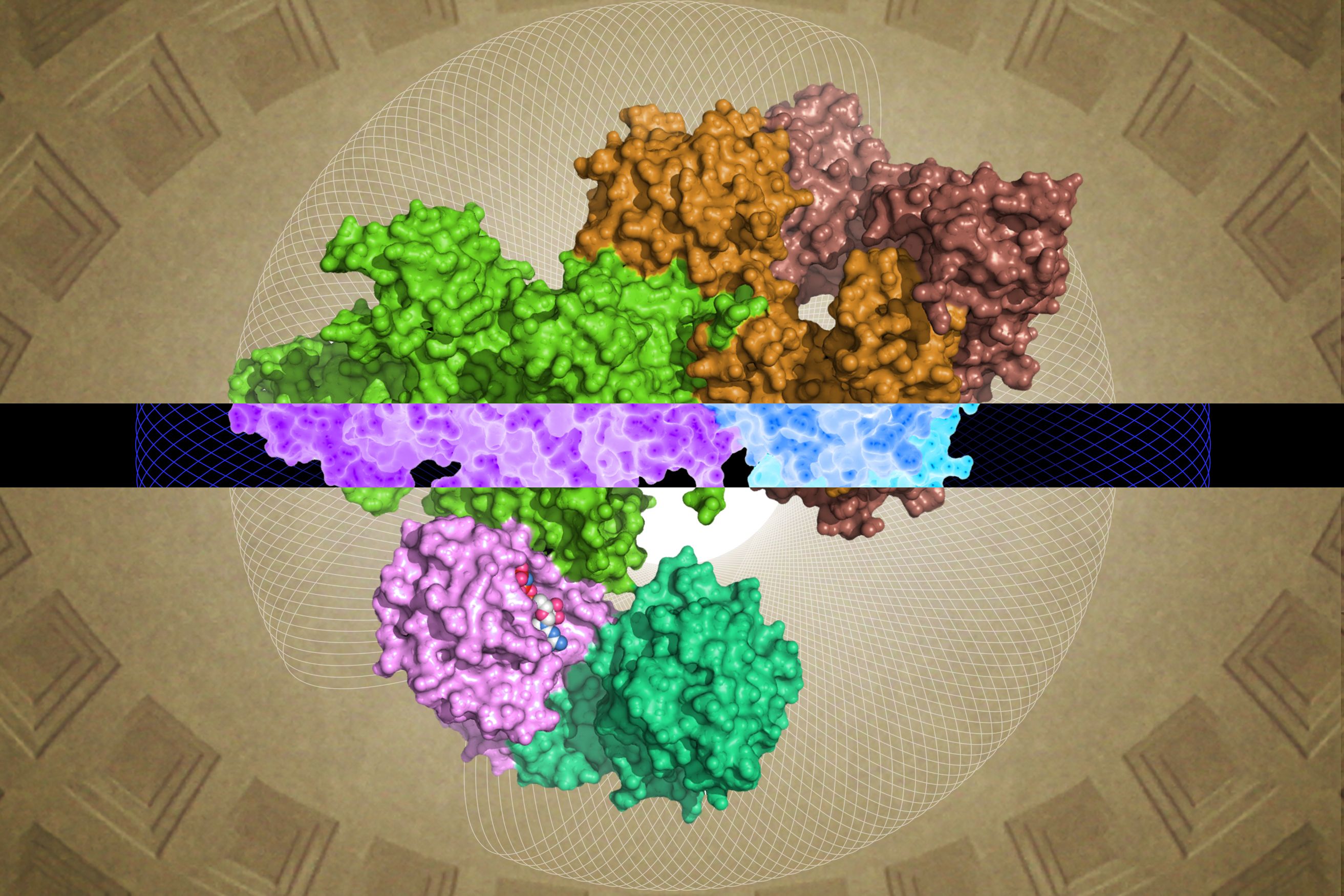
The study of the relative amounts of substances — including proteins — is known as “stoichiometry.” To investigate the stoichiometry of enzymes in different types of cells, Li and his colleagues analyzed three different species of bacteria — Escherichia coli, Bacillus subtilis, and Vibrio natriegens — as well as the budding yeast Saccharomyces cerevisiae. Among these cells, scientists compared the amount of enzymes in 21 pathways responsible for a variety of tasks including repairing DNA, constructing fatty acids, and converting sugar to energy. Because these species of yeast and bacteria have evolved to live in different environments and have different cellular structures, such as the presence or lack of a nucleus, researchers were surprised to find that all four cells types had nearly identical enzyme stoichiometry in all pathways examined.
Li’s team followed up their unexpected results by detailing how bacteria achieve a consistent enzyme stoichiometry. Cells control enzyme production by regulating two processes. The first, transcription, converts the information contained in a strand of DNA into many copies of messenger RNA (mRNA). The second, translation, occurs as ribosomes decode the mRNAs to construct proteins. By analyzing transcription across all three bacterial species, Li’s team discovered that the different bacteria produced varying amounts of mRNA encoding for enzymes in a pathway.
Different amounts of mRNA theoretically lead to differences in protein production, but the researchers found instead that the cells adjusted their rates of translation to compensate for changes in transcription. Cells that produced more mRNA slowed their rates of protein synthesis, while cells that produced less mRNA increased the speed of protein synthesis. Thanks to this compensation, the stoichiometry of enzymes remained constant across the different bacteria.
“It is remarkable that E. coli and B. subtilis need the same relative amount of the corresponding proteins, as seen by the compensatory variations in transcription and translation efficiencies,” says Johan Elf, professor of physical biology at Uppsala University in Sweden. “These results raise interesting questions about how enzyme production in different cells have evolved.”
“Examining bacterial gene clusters was really striking,” lead author Lalanne says. “Over a long evolutionary history, these genes have shifted positions, mutated into different sequences, and been bombarded by mobile pieces of DNA that randomly insert themselves into the genome. Despite all this, the bacteria have compensated for these changes by adjusting translation to maintain the stoichiometry of their enzymes. This suggests that evolutionary forces, which we don’t yet understand, have shaped cells to have the same enzyme stoichiometry.”
Searching for the stoichiometry regulating human health
In the future, Li and his colleagues will test whether their findings in bacteria and yeast extend to humans. Because unicellular and multicellular organisms manage energy and nutrients differently, and experience different selection pressures, researchers are not sure what they will discover.
“Perhaps there will be enzymes whose stoichiometry varies, and a smaller subset of enzymes whose levels are more conserved,” Li says. “This would indicate that the human body is sensitive to changes in specific enzymes that could make good drug targets. But we won’t know until we look.”
Beyond the human body, Li and his team believe that it is possible to find simplicity underlying the complex bustle of molecules within all cells. Like other mathematical patterns in nature, such as the the spiral of seashells or the branching pattern of trees, the stoichiometry of enzymes may be a widespread design principle of life.
The research was funded by the National Institutes of Health, Pew Biomedical Scholars Program, Sloan Research Fellowship, Searle Scholars Program, National Sciences and Engineering Research Council of Canada, Howard Hughes Medical Institute, National Science Foundation, Helen Hay Whitney Foundation, Jane Coffin Childs Memorial Fund, and the Smith Family Foundation.