
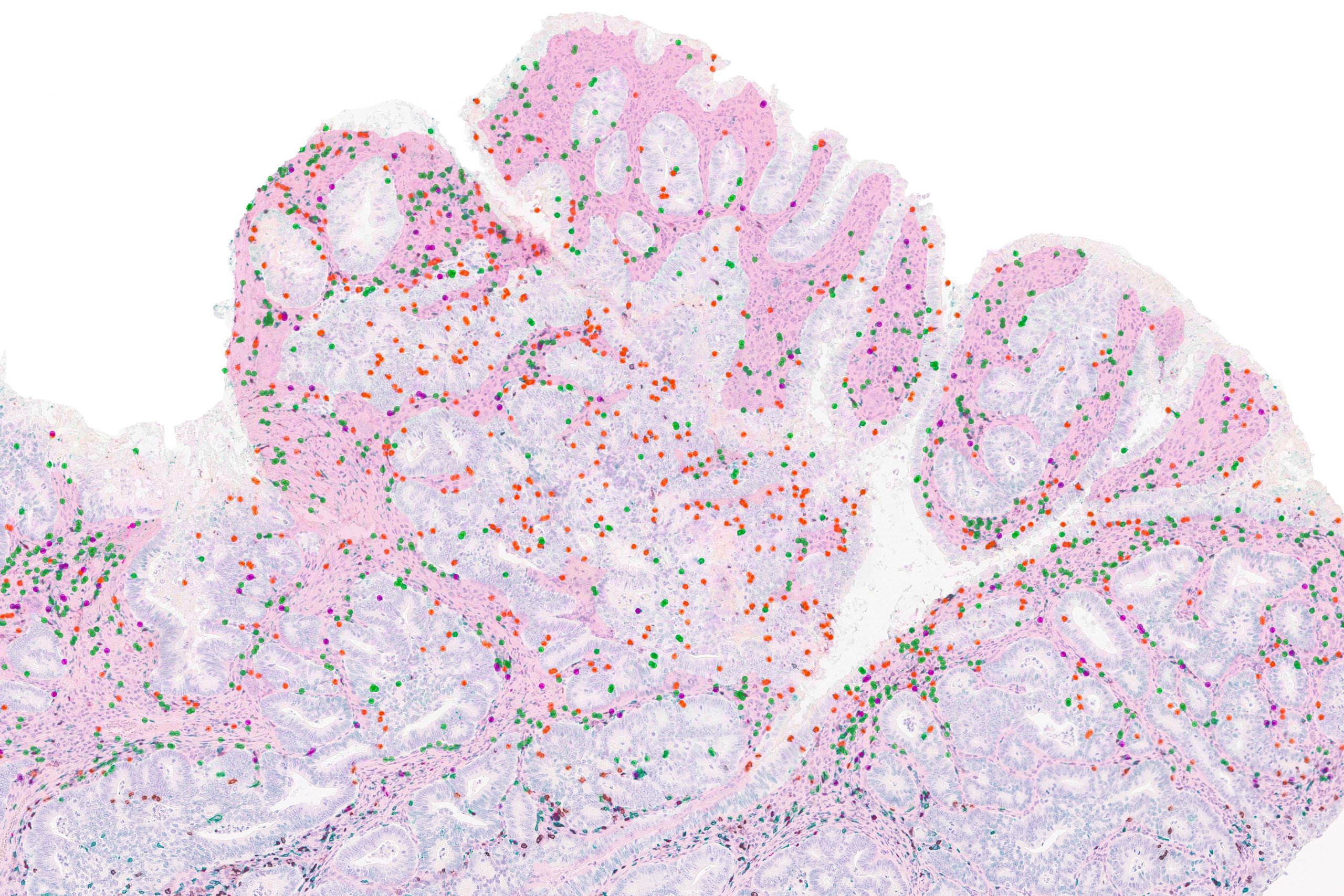
Cancer drugs known as checkpoint blockade inhibitors have proven effective for some cancer patients. These drugs work by taking the brakes off the body’s T cell response, stimulating those immune cells to destroy tumors.
Some studies have shown that these drugs work better in patients whose tumors have a very large number of mutated proteins, which scientists believe is because those proteins offer plentiful targets for T cells to attack. However, for at least 50 percent of patients whose tumors show a high mutational burden, checkpoint blockade inhibitors don’t work at all.
A new study from MIT reveals a possible explanation for why that is. In a study of mice, the researchers found that measuring the diversity of mutations within a tumor generated much more accurate predictions of whether the treatment would succeed than measuring the overall number of mutations.
If validated in clinical trials, this information could help doctors to better determine which patients will benefit from checkpoint blockade inhibitors.
“While very powerful in the right settings, immune checkpoint therapies are not effective for all cancer patients. This work makes clear the role of genetic heterogeneity in cancer in determining the effectiveness of these treatments,” says Tyler Jacks, the David H. Koch Professor of Biology and a member of MIT’s Koch Institute for Cancer Research.
Jacks; Peter Westcott, a former MIT postdoc in the Jacks lab who is now an assistant professor at Cold Spring Harbor Laboratory; and Isidro Cortes-Ciriano, a research group leader at EMBL’s European Bioinformatics Institute (EMBL-EBI), are the senior authors of the paper, which appears today in Nature Genetics.
A diversity of mutations
Across all types of cancer, a small percentage of tumors have what is called a high tumor mutational burden (TMB), meaning they have a very large number of mutations in each cell. A subset of these tumors has defects related to DNA repair, most commonly in a repair system known as DNA mismatch repair.
Because these tumors have so many mutated proteins, they are believed to be good candidates for immunotherapy treatment, as they offer a plethora of potential targets for T cells to attack. Over the past few years, the FDA has approved a checkpoint blockade inhibitor called pembrolizumab, which activates T cells by blocking a protein called PD-1, to treat several types of tumors that have a high TMB.
However, subsequent studies of patients who received this drug found that more than half of them did not respond well or only showed short-lived responses, even though their tumors had a high mutational burden. The MIT team set out to explore why some patients respond better than others, by designing mouse models that closely mimic the progression of tumors with high TMB.
These mouse models carry mutations in genes that drive cancer development in the colon and lung, as well as a mutation that shuts down the DNA mismatch repair system in these tumors as they begin to develop. This causes the tumors to generate many additional mutations. When the researchers treated these mice with checkpoint blockade inhibitors, they were surprised to find that none of them responded well to the treatment.
“We verified that we were very efficiently inactivating the DNA repair pathway, resulting in lots of mutations. The tumors looked just like they look in human cancers, but they were not more infiltrated by T cells, and they were not responding to immunotherapy,” Westcott says.
The researchers discovered that this lack of response appears to be the result of a phenomenon known as intratumoral heterogeneity. This means that, while the tumors have many mutations, each cell in the tumor tends to have different mutations than most of the other cells. As a result, each individual cancer mutation is “subclonal,” meaning that it is expressed in a minority of cells. (A “clonal” mutation is one that is expressed in all of the cells.)
In further experiments, the researchers explored what happened as they changed the heterogeneity of lung tumors in mice. They found that in tumors with clonal mutations, checkpoint blockade inhibitors were very effective. However, as they increased the heterogeneity by mixing tumor cells with different mutations, they found that the treatment became less effective.
“That shows us that intratumoral heterogeneity is actually confounding the immune response, and you really only get the strong immune checkpoint blockade responses when you have a clonal tumor,” Westcott says.
Failure to activate
It appears that this weak T cell response occurs because the T cells simply don’t see enough of any particular cancerous protein, or antigen, to become activated, the researchers say. When the researchers implanted mice with tumors that contained subclonal levels of proteins that normally induce a strong immune response, the T cells failed to become powerful enough to attack the tumor.
“You can have these potently immunogenic tumor cells that otherwise should lead to a profound T cell response, but at this low clonal fraction, they completely go stealth, and the immune system fails to recognize them,” Westcott says. “There’s not enough of the antigen that the T cells recognize, so they’re insufficiently primed and don’t acquire the ability to kill tumor cells.”
To see if these findings might extend to human patients, the researchers analyzed data from two small clinical trials of people who had been treated with checkpoint blockade inhibitors for either colorectal or stomach cancer. After analyzing the sequences of the patients’ tumors, they found that patients’ whose tumors were more homogeneous responded better to the treatment.
“Our understanding of cancer is improving all the time, and this translates into better patient outcomes,” Cortes-Ciriano says. “Survival rates following a cancer diagnosis have significantly improved in the past 20 years, thanks to advanced research and clinical studies. We know that each patient’s cancer is different and will require a tailored approach. Personalized medicine must take into account new research that is helping us understand why cancer treatments work for some patients but not all.”
The findings also suggest that treating patients with drugs that block the DNA mismatch repair pathway, in hopes of generating more mutations that T cells could target, may not help and could be harmful, the researchers say. One such drug is now in clinical trials.
“If you try to mutate an existing cancer, where you already have many cancer cells at the primary site and others that may have disseminated throughout the body, you’re going to create a super heterogeneous collection of cancer genomes. And what we showed is that with this high intratumoral heterogeneity, the T cell response is confused and there is absolutely no response to immune checkpoint therapy,” Westcott says.
The research was funded by the Koch Institute Support (core) Grant from the U.S. National Cancer Institute, the Howard Hughes Medical Institute, and a Damon Runyon Fellowship Award.
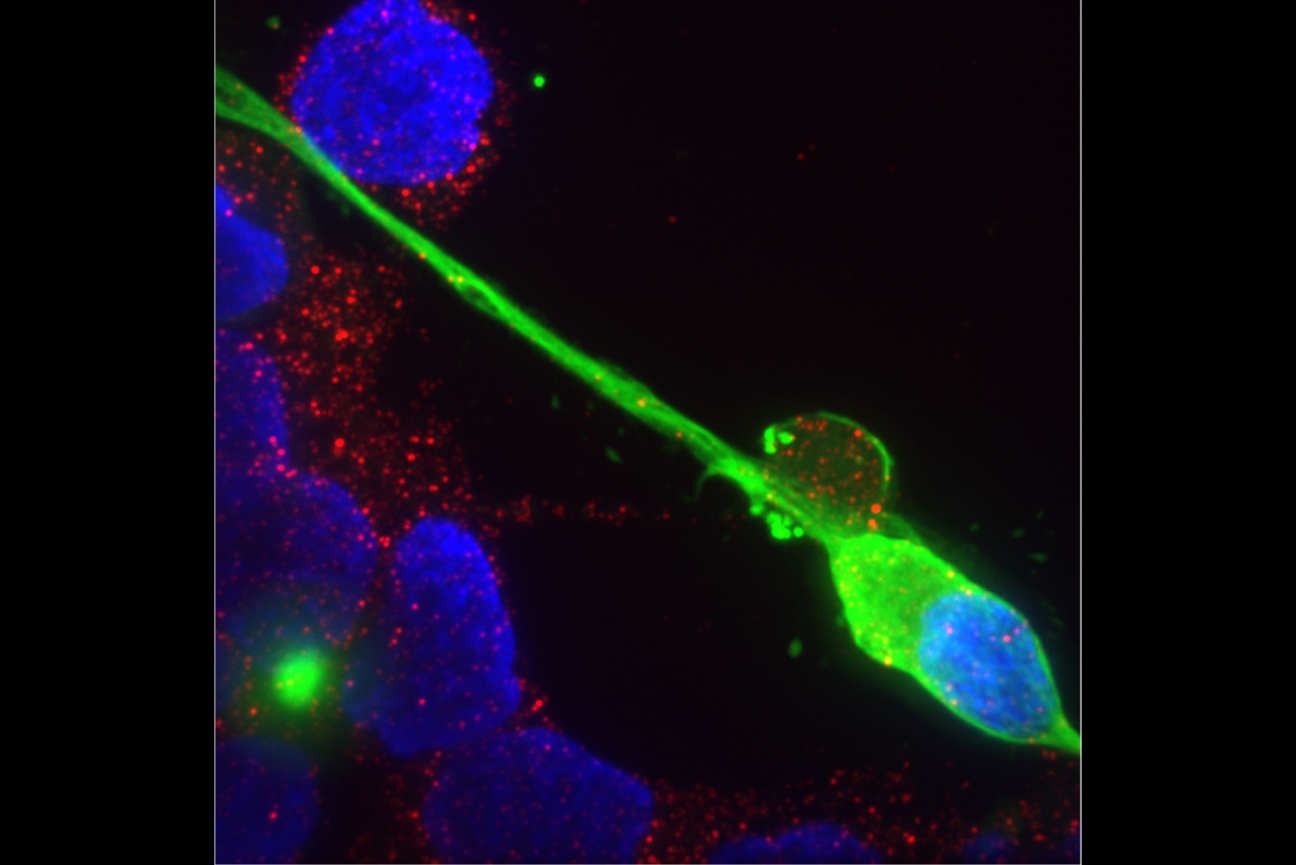
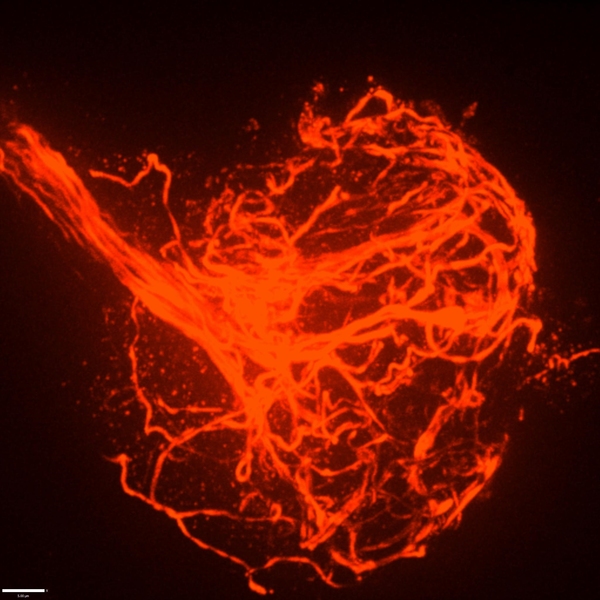
Perhaps the most obvious feature of a neuron is the long branch called an axon that ventures far from the cell body to connect with other neurons or muscles. If that long, thin projection ever seems like it could be vulnerable, a new MIT study shows that its structural integrity may indeed require the support of a surrounding protein called perlecan. Without that protein in Drosophila fruit flies, researchers at The Picower Institute for Learning and Memory found axonal segments can break apart during development and the connections, or synapses, that they form end up dying away.
Perlecan helps make the extracellular matrix, the proteins and other molecules that surround cells, stable and flexible so that cells can develop and function in an environment that is supportive without being rigid.
“What we found was that the extracellular matrix around nerves was being altered and essentially causing the nerves to break completely. Broken nerves eventually led to the synapses retracting,” says study senior author Troy Littleton, the Menicon Professor in MIT’s departments of Biology and Brain and Cognitive Sciences.
Humans need at least some perlecan to survive after birth. Mutations that reduce, but don’t eliminate, perlecan can cause Schwartz-Jampel syndrome, in which patients experience neuromuscular problems and skeletal abnormalities. The new study may help explain how neurons are affected in the condition, Littleton says, and also deepen scientists’ understanding of how the extracellular matrix supports axon and neural circuit development.
Ellen Guss PhD ’23, who recently defended her doctoral thesis on the work, led the research published June 8 in eLife.
At first she and Littleton didn’t expect the study to yield a new discovery about the durability of developing axons. Instead, they were investigating a hypothesis that perlecan might help organize some of the protein components in synapses that fly nerves develop to connect with muscles. But when they knocked out the gene called “trol” that encodes perlecan in flies, they saw that the neurons appeared to “retract” many synapses at a late stage of larval development. Proteins on the muscle side of the synaptic connection remained, but the neuron side of the connection withered away. That suggested that perlecan had a bigger role than they first thought.
Indeed, the authors found that the perlecan wasn’t particularly enriched around synapses. Where it was pronounced was in a structure called the neural lamella, which surrounds axon bundles and acts a bit like the rubbery cladding around a TV cable to keep the structure intact. That suggested that a lack of perlecan might not be a problem at the synapse, but instead causes trouble along axons due to its absence in the extracellular matrix surrounding nerve bundles.
Littleton’s lab had developed a technique for daily imaging of fly neural development called serial intravital imaging. They applied it to watch what happened to the fly axons and synapses over a four-day span. They observed that while fly axons and synapses developed normally at first, not only synapses but also whole segments of axons faded away.
They also saw that the farther an axon segment was from the fly’s brain, the more likely it was to break apart, suggesting that the axon segments became more vulnerable the further out they extended. Looking segment by segment, they found that where axons were breaking down, synapse loss would soon follow, suggesting that axon breakage was the cause of the synapse retraction.
“The breakages were happening in a segment-wide manner,” Littleton says. “In some segments the nerves would break and in some they wouldn’t. Whenever there was a breakage event, you would see all the neuromuscular junctions (synapses) across all the muscles in that segment retract.”
When they compared the structure of the lamella in mutant versus healthy flies, they found that the lamella was thinner and defective in the mutants. Moreover, where the lamella was weakened, axons were prone to break and the microtubule structures that run the length of the axon would become misdirected, protruding outward and becoming tangled up in dramatic bundles at sites of severed axons.
In one other key finding, the team showed that perlecan’s critical role depended on its secretion from many cells, not just neurons. Blocking the protein in just one cell type or another did not cause the problems that total knockdown did, and enhancing secretion from just neurons was not enough to overcome its deficiency from other sources.
Altogether, the evidence pointed to a scenario where lack of perlecan secretion caused the neural lamella to be thin and defective, with the extracellular matrix becoming too rigid. The further from the brain nerve bundles extended, the more likely movement stresses would cause the axons to break where the lamella had broken down. The microtubule structure within the axons then became disorganized. That ultimately led to synapses downstream of those breakages dying away because the disruption of the microtubules means the cells could no longer support the synapses.
“When you don’t have that flexibility, although the extracellular matrix is still there, it becomes very rigid and tight and that basically leads to this breakage as the animal moves and pulls on those nerves over time,” Littleton says. “It argues that the extracellular matrix is functional early on and can support development, but doesn’t have the right properties to sustain some key functions over time as the animal begins to move and navigate around. The loss of flexibility becomes really critical.”
In addition to Littleton and Guss, the paper’s other authors are Yulia Akbergenova and Karen Cunningham.
Support for the study came from the National Institutes of Health. The Littleton Lab is also supported by The Picower Institute for Learning and Memory and The JPB Foundation.

There were early signs that Nicole De Nisco, SB ‘07, PhD ‘13, might become a scientist. She ran out of science classes to take in high school and fondly remembers the teacher that encouraged her to pursue science instead of the humanities. But she ended up at MIT, in part, out of spite.
“I applied because my guidance counselor told me I wouldn’t get in,” she said. The rest, as they say, is history for the first-generation college student from Los Angeles.
Now, she’s an assistant professor of biological sciences at UT Dallas studying urinary tract infections (UTIs) and the urinary microbiome in postmenopausal women.
De Nisco has already made some important advancements in the field: she developed a new technique for visualizing bacteria in the bladder and used it to demonstrate that bacteria form reservoirs in human bladder tissue, leading to chronic or recurrent UTIs.
It was known that in mice, bacteria are able to create communities within the bladder tissue, forming reservoirs and staying there long term—but no one had shown that occurring in human tissue before.

De Nisco found that reservoirs of tissue-resident bacteria exist in human patients with recurring UTIs, a condition which may ultimately lead to women needing to have their bladder removed. De Nisco now mostly works with postmenopausal women who have been suffering from decades of recurring UTIs.
There was a big gap in the field, De Nisco explained, so entering the field of urology was also an opportunity to make new discoveries and find new ways to treat those recurring infections.
De Nisco said she’s in the minority, both as a woman studying urology and as someone studying diseases that affect female patients. Most researchers in the urology field are men, and most focus on the prostate.
But things are changing.
“I think there are a lot of women in the field who are now pushing back, and I actually collaborate with a lot of other female investigators in the field. We’re trying to support each other so that we can survive and, hopefully, actually advance the science—instead of it being in the same place it was 15 years ago,” De Nisco says.
De Nisco first fell in love with biomedical research as an undergrad doing a UROP in Catherine Drennan’s lab, back when Drennan was still located in the chemistry building.
“Cathy herself was incredibly encouraging, and is probably the main reason I decided to pursue a career in science—or felt that I could,” De Nisco said.
De Nisco became fascinated with the dialogue between a microbe and a host organism during an undergraduate course in microbial physiology with Graham Walker, which led to De Nisco’s decision to remain at MIT for her PhD work and to perform her doctoral research in rhizobia legume symbiosis in Walker’s lab.
De Nisco said during her time at MIT, Drennan and Walker gave her a lot of encouragement and “room to do my own thing,” fostering a love of discovery and problem solving. It’s a mentoring style she’s using now with her own graduate students; she currently has eight working in her lab.
“Every student is different: some just want a project and they want to know what they’re doing, and some want to explore,” she said. “I was the type that wanted to do my own thing and so they gave me the room and the patience to be able to explore and find something new that I was interested in and excited about.”
As a low-income student sending financial help home, she also pursued teaching opportunities outside of her usual duties; Walker was very supportive of pursuing other teaching opportunities. De Nisco was a graduate student tutor for Next House watching over 40 undergrads, served as a teaching fellow with the Harvard Extension School, and worked with Eric Lander to help launch the course 7.00x Introduction to Biology – The Secret of Life for EdX, one of the most highly rated MOOCs of all time.
She said MIT definitely prepared her for a life as a professor, teacher, and mentor; the most important thing about graduate school isn’t choosing “the most cutting-edge research project,” but making sure you have a good training experience and an advisor who can provide that.
“You don’t need to start building your name in the field when you’re a grad student. The lab environment is much more important than the topic. It’s easy to get burned out or to be turned off to a career in academia altogether if you have the wrong advisor,” she said. “You need to learn how to be a scientist, and you have plenty of time later in your career to follow whatever path you want to follow.”
She knows this from experience: her current research is somewhat parallel but unrelated to her previous research experience.
“I think my motivation for being a scientist is rooted in my desire to help people doing something I enjoy,” she said. “I was not doing this kind of research as a graduate student, and that doesn’t mean that I wasn’t able to end up at this point in my career where I’m doing research that is focused on improving the lives of women, specifically.”
She did her postdoctoral work at UT Southwestern Medical Center studying Vibrio parahaemolyticus, a human pathogen that causes gastroenteritis. The work was a marriage of her interests in biochemistry and host-microbiome interactions.
She said MIT prepared her well for the type of interdisciplinary work that she does every day: At UT Dallas, all the research buildings are fully integrated, with engineers, chemists, physicists, and biologists sharing lab spaces in the same building. Her closest collaborators are mathematicians, chemists, and engineers.
Although she may not be fully literate, she has a common language with the people she works with thanks to MIT’s undergraduate course requirements in many different topics and MIT’s focus on interdisciplinary research, which is “how real advancement is made.”
Ultimately, De Nisco said she is glad to this day that she attended MIT.
“Getting that acceptance letter to attend MIT probably changed the trajectory of my life,” she said. “You never know, on paper, what someone is going to achieve eventually, and what kind of force they’re going to be. I’m always grateful to whoever was on the admissions committees that made the decision to accept me—twice.”

Many of our body’s most important functions occur without our conscious knowledge, such as digestion, heartbeat, and breathing. These vital functions depend on the signals generated by the “interoceptive nervous system,” which enables the brain to monitor our internal organs and trigger responses that sometimes save our lives. One second you are breathing normally as you eat your salad and the next, when a vinegar-soaked crouton enters your throat, you are coughing or swallowing to protect and clear your airway. We know our bodies are sensitive to cues like irritants, but we still have a lot to learn about how the interoceptive system works to meet our physiological needs, keep organs safe and healthy, and affect our behavior. We can also learn how chronic insults may lead to organ dysfunction and use what we learn to create therapeutic interventions.
Focusing on the airway, Sara Prescott, a new faculty member in the Department of Biology and investigator in The Picower Institute for Learning and Memory, seeks to understand the ways our nervous systems detect and respond to stimuli in health and disease. Here, she describes her work.
Q: Why is understanding the peripheral nervous system important, and what parts of your background are you drawing on for your current research?
A: The lab focuses on really trying to explore the body-brain connection.
People often think that our mind exists in a vacuum, but in reality, our nervous system is heavily integrated with the rest of the body, and those neural interfaces are important, both for taking information from our body or environment and turning it into an internal representation of the world, and, in reverse, being able to process that information and being able to enact changes throughout the body. That includes things like autonomic reflexes, basic functions of the body like breathing, blood-gas regulation, digestion, and heart rate.
I’ve integrated both my graduate training and postdoctoral training into thinking about biology across multiple scales.
Graduate school for me was quite focused on deep molecular mechanism questions, particularly gene regulation, so I feel like that has been very useful for me in my general approach to neuroscience because I take a very molecular angle to all of this.
It also showed me the power of in vitro models as reductionist tools to explore fundamental aspects of cell biology. During my postdoc, I focused on larger, emergent phenotypes. We were able to manipulate specific circuits and see very impressive behavioral responses in animals. You could stimulate about 100 neurons in a mouse and see that their breathing would just stop until you remove the stimulation, and then the breathing would return to normal.
Both of those experiences inform how we approach a problem in my research. We need to understand how these circuits work, not just their connectivity at the anatomical level but what is driving their changes in sensitivity over time, the receptor expression programs that affect how they sense and signal, how these circuits emerge during development, and their gene expression.
There are still so many foundational questions that haven’t been answered that there’s enough to do in the mouse for quite some time.
Q: How are you specifically looking into interoceptive biology at MIT?
A: Our flagship system is the mammalian airway. We use a mouse model and modern molecular neuroscience tools to manipulate various neural pathways and observe what the effects are on respiratory function and animal health.
Neuroscience and mouse work have a reputation for being a little challenging and intense, but I think this is also where we can ask really important questions that are useful for our everyday lives — and the only place where we can fully recapitulate the complexity of nervous system signaling all the way down to our organs, back to our brain, and back to our organs.
It’s a very fun place to do science with lots of open questions.
One of the core discoveries from my postdoctoral work was focusing on the vagus nerve as a major body-to-brain conduit, as it innervates our lungs, heart, and gastrointestinal tract. We found that there were about 40 different subtypes of sensory neurons within this small nerve, which is really a remarkable amount of diversity and reflects the massive sensory space within the body. About a dozen of those vagal neurons project to the airways.
We identified a rare neuron type specifically responsible for triggering protective responses, like coughing when water or acid entered the airway. We also discovered a separate population of neurons that make us feel and act sick when we get a flu infection. The field now knows what four to five vagal populations of neurons are actually sensing in the airways, but the remaining populations are still a mystery to us; we don’t know what those populations of sensory neurons are detecting, what their anatomy is, and what reflex effects those neurons are evoking.
Looking ahead, there are many exciting directions for the interoceptive biology field. For example, there’s been a lot of focus on characterizing the circuits underlying acute motor reflexes, like rapid responses to visceral stimuli on the timescale of minutes to hours. But we don’t have a lot of information about what happens when these circuits are activated over long periods of time. For example, respiratory tract infections often last for weeks or longer. We know that the airways undergo changes in composition when they’re exposed to different types of infection or stress to better accommodate future threats. One of the hypotheses we’re testing is that chronically activating neural circuits may drive changes in organ composition. We have this idea, which we’re calling reflexive remodeling: neurons may be communicating with stem cells and progenitor cells in the periphery to drive adaptive remodeling responses.
We have the genetic, molecular, and circuit scale tools to explore this phenomenon in mice. In parallel, we’re also setting up some in vitro models of the mouse airway mucosa to expedite receptor screening and to explore basic mechanisms of neuron-epithelium cross-talk. We hope this will inform our understanding of how the airway surface senses and responds to different types of irritants or damage.
Q: This all sounds fascinating. Where does it lead?
A: Human health has been my north star for a long time and I’ve taken a long, wandering path to find particular areas where I can scratch whatever intellectual itch that I have.
I originally thought I would be a doctor and then realized that I felt like I could have a more lasting impact by discovering fundamental truths about how our bodies work. I think there are a number of chronic diseases in which autonomic imbalance is actually a huge clinical component of the disorder.
We have a lot of interest in some of these very common airway remodeling diseases, like chronic obstructive pulmonary disorder — COPD — asthma, and potentially lung cancer. We want to ask questions like how autonomic circuits are altered in disease contexts, and when neurons actually drive features of disease.
Perhaps this research will help us come up with better molecular, cellular, or tissue engineering approaches to improve the outcomes for a variety of autonomic diseases.
It’s very easy for me to imagine how one day, not too far from now, we can turn these findings into something actionable for human health.
Genomic studies of cancer patients have revealed thousands of mutations linked to tumor development. However, for the vast majority of those mutations, researchers are unsure of how they contribute to cancer because there’s no easy way to study them in animal models.
In an advance that could help scientists make a dent in that long list of unexplored mutations, MIT researchers have developed a way to easily engineer specific cancer-linked mutations into mouse models.
Using this technique, which is based on CRISPR genome-editing technology, the researchers have created models of several different mutations of the cancer-causing gene Kras, in different organs. They believe this technique could also be used for nearly any other type of cancer mutation that has been identified.
Such models could help researchers identify and test new drugs that target these mutations.
“This is a remarkably powerful tool for examining the effects of essentially any mutation of interest in an intact animal, and in a fraction of the time required for earlier methods,” says Tyler Jacks, the David H. Koch Professor of Biology, a member of the Koch Institute for Integrative Cancer Research at MIT, and one of the senior authors of the new study.
Francisco Sánchez-Rivera, an assistant professor of biology at MIT and member of the Koch Institute, and David Liu, a professor in the Harvard University Department of Chemistry and Chemical Biology and a core institute member of the Broad Institute, are also senior authors of the study, which appears today in Nature Biotechnology.
Zack Ely PhD ’22, a former MIT graduate student who is now a visiting scientist at MIT, and MIT graduate student Nicolas Mathey-Andrews are the lead authors of the paper.
Faster editing
Testing cancer drugs in mouse models is an important step in determining whether they are safe and effective enough to go into human clinical trials. Over the past 20 years, researchers have used genetic engineering to create mouse models by deleting tumor suppressor genes or activating cancer-promoting genes. However, this approach is labor-intensive and requires several months or even years to produce and analyze mice with a single cancer-linked mutation.
“A graduate student can build a whole PhD around building a model for one mutation,” Ely says. “With traditional models, it would take the field decades to catch up to all of the mutations we’ve discovered with the Cancer Genome Atlas.”
In the mid-2010s, researchers began exploring the possibility of using the CRISPR genome-editing system to make cancerous mutations more easily. Some of this work occurred in Jacks’ lab, where Sánchez-Rivera (then an MIT graduate student) and his colleagues showed that they could use CRISPR to quickly and easily knock out genes that are often lost in tumors. However, while this approach makes it easy to knock out genes, it doesn’t lend itself to inserting new mutations into a gene because it relies on the cell’s DNA repair mechanisms, which tend to introduce errors.
Inspired by research from Liu’s lab at the Broad Institute, the MIT team wanted to come up with a way to perform more precise gene-editing that would allow them to make very targeted mutations to either oncogenes (genes that drive cancer) or tumor suppressors.
In 2019, Liu and colleagues reported a new version of CRISPR genome-editing called prime editing. Unlike the original version of CRISPR, which uses an enzyme called Cas9 to create double-stranded breaks in DNA, prime editing uses a modified enzyme called Cas9 nickase, which is fused to another enzyme called reverse transcriptase. This fusion enzyme cuts only one strand of the DNA helix, which avoids introducing double-stranded DNA breaks that can lead to errors when the cell repairs the DNA.
The MIT researchers designed their new mouse models by engineering the gene for the prime editor enzyme into the germline cells of the mice, which means that it will be present in every cell of the organism. The encoded prime editor enzyme allows cells to copy an RNA sequence into DNA that is incorporated into the genome. However, the prime editor gene remains silent until activated by the delivery of a specific protein called Cre recombinase.
Since the prime editing system is installed in the mouse genome, researchers can initiate tumor growth by injecting Cre recombinase into the tissue where they want a cancer mutation to be expressed, along with a guide RNA that directs Cas9 nickase to make a specific edit in the cells’ genome. The RNA guide can be designed to induce single DNA base substitutions, deletions, or additions in a specified gene, allowing the researchers to create any cancer mutation they wish.
Modeling mutations
To demonstrate the potential of this technique, the researchers engineered several different mutations into the Kras gene, which drives about 30 percent of all human cancers, including nearly all pancreatic adenocarcinomas. However, not all Kras mutations are identical. Many Kras mutations occur at a location known as G12, where the amino acid glycine is found, and depending on the mutation, this glycine can be converted into one of several different amino acids.
The researchers developed models of four different types of Kras mutations found in lung cancer: G12C, G12D, G12R, and G12A. To their surprise, they found that the tumors generated in each of these models had very different traits. For example, G12R mutations produced large, aggressive lung tumors, while G12A tumors were smaller and progressed more slowly.
Learning more about how these mutations affect tumor development differently could help researchers develop drugs that target each of the different mutations. Currently, there are only two FDA-approved drugs that target Kras mutations, and they are both specific to the G12C mutation, which accounts for about 30 percent of the Kras mutations seen in lung cancer.
The researchers also used their technique to create pancreatic organoids with several different types of mutations in the tumor suppressor gene p53, and they are now developing mouse models of these mutations. They are also working on generating models of additional Kras mutations, along with other mutations that help to confer resistance to Kras inhibitors.
“One thing that we’re excited about is looking at combinations of mutations including Kras mutations that drives tumorigenesis, along with resistance associated mutations,” Mathey-Andrews says. “We hope that will give us a handle on not just whether the mutation causes resistance, but what does a resistant tumor look like?”
The researchers have made mice with the prime editing system engineered into their genome available through a repository at the Jackson Laboratory, and they hope that other labs will begin to use this technique for their own studies of cancer mutations.
The research was funded by the Ludwig Center at MIT, the National Cancer Institute, a Howard Hughes Medical Institute Hanna Grey Fellowship, the V Foundation for Cancer Research, a Koch Institute Frontier Award, the MIT Research Support Committee, a Helen Hay Whitney Postdoctoral Fellowship, the David H. Koch Graduate Fellowship Fund, the National Institutes of Health, and the Lustgarten Foundation for Pancreatic Cancer Research.
Other authors of the paper include Santiago Naranjo, Samuel Gould, Kim Mercer, Gregory Newby, Christina Cabana, William Rideout, Grissel Cervantes Jaramillo, Jennifer Khirallah, Katie Holland, Peyton Randolph, William Freed-Pastor, Jessie Davis, Zachary Kulstad, Peter Westcott, Lin Lin, Andrew Anzalone, Brendan Horton, Nimisha Pattada, Sean-Luc Shanahan, Zhongfeng Ye, Stefani Spranger, and Qiaobing Xu.

Two faculty members from the MIT Department of Biology have been selected by the Howard Hughes Medical Institute (HHMI) for the inaugural cohort of HHMI Freeman Hrabowski Scholars.
Seychelle Vos, the Robert A. Swanson Career Development Professor of Life Sciences, and Hernandez Moura Silva, an assistant professor of biology and core member of the Ragon Institute of MGH, MIT and Harvard, are among 31 early-career faculty selected for their potential to become leaders in their research fields and to create diverse and inclusive lab environments in which everyone can thrive, according to a press release.
Freeman Hrabowski Scholars are appointed to a five-year term, renewable for a second five-year term after a successful progress evaluation. Each scholar will receive up to $8.6 million over 10 years, including full salary, benefits, a research budget, and scientific equipment. In addition, they will participate in professional development to advance their leadership and mentorship skills.
The Freeman Hrabowski Scholars Program represents a key component of HHMI’s diversity, equity, and inclusion goals. Over the next 20 years, HHMI expects to hire and support up to 150 Freeman Hrabowski Scholars — appointing roughly 30 scholars every other year for the next 10 years. The institute has committed up to $1.5 billion for the Freeman Hrabowski Scholars to be selected over the next decade. The program was named for Freeman A. Hrabowski III, president emeritus of the University of Maryland at Baltimore County, who played a major role in increasing the number of scientists, engineers, and physicians from backgrounds underrepresented in science in the United States.
Seychelle Vos
Seychelle Vos studies how DNA organization impacts gene expression at the atomic level, using cryogenic electron microscopy (cryo-EM), X-ray crystallography, biochemistry, and genetics. Human cells contain about 2 meters of DNA, which is packed so tightly that its entirety is contained within the nucleus, which is only a few microns across. Although DNA needs to be compacted, it also needs to be accessible to, and readable by, the cell’s molecular machinery.
Vos received a BS in genetics from the University of Georgia in 2008 and a PhD from University of California at Berkeley in 2013. During her postdoctoral research at the Max Planck Institute for Biophysical Chemistry in Germany, she determined how the molecular machine responsible for gene expression is regulated near gene promoters.
Vos joined MIT as an assistant professor of biology in fall 2019.
“I am very humbled and honored to have been named a HHMI Freeman Hrabowski Scholar,” Vos says. “It would not have been possible without the hard work of my lab and the help of my colleagues. It provides us with the support to achieve our ambitious research goals.”
Hernandez Moura Silva
Hernandez Moura Silva studies the role of immune cells in the maintenance and normal function of our bodies and tissues, beyond their role in battling infection. Specifically, he looks at a specific type of immune cell called a macrophage and its role in the proper function of white adipose tissue — our fat. White adipose tissue in a healthy state is highly populated by macrophages, including very abundant ones known as “vasculature-associated adipose tissue macrophages,” which are located around the blood vessels. When the activity of these adipose macrophages is disrupted, there are changes in the proper function of the white adipose tissue, which may ultimately link to disease. By understanding macrophage function in healthy tissues, Hernandez hopes to learn how to restore tissue homeostasis in disease.
Hernandez Moura Silva received a BS in biology in 2005 and an MSc in molecular biology in 2008 from the University of Brazil. He received his PhD in 2011 from the University of São Paulo Heart Institute. Silva pursued his postdoctoral work as the Bernard Levine Postdoctoral Fellow in immunology and immuno-metabolism at the New York University School of Medicine Skirball Institute of Biomolecular Medicine.
He joined MIT as an assistant professor of biology in 2022. He is also a core member of the Ragon Institute.
“For an immigrant coming from an underrepresented group, it’s a huge privilege to be granted this opportunity from HHMI that will empower me and my lab to shape the next generation of scientists and provide an environment where people can feel welcome and encouraged to do the science that they love and be successful,” Silva says. “It also aligns with MIT’s commitment to increase diversity and opportunity across the Institute and to become a place where all people can thrive.”

Many of our body’s most important functions occur without our conscious knowledge, such as digestion, heartbeat, and breathing. These vital functions depend on the signals generated by the “interoceptive nervous system,” which enables the brain to monitor our internal organs and trigger responses that sometimes save our lives. One second you are breathing normally as you eat your salad and the next, when a vinegar-soaked crouton enters your throat, you are coughing or swallowing to protect and clear your airway. We know our bodies are sensitive to cues like irritants, but we still have a lot to learn about how the interoceptive system works to meet our physiological needs, keep organs safe and healthy, and affect our behavior. We can also learn how chronic insults may lead to organ dysfunction and use what we learn to create therapeutic interventions.
Focusing on the airway, Sara Prescott, a new faculty member in the Department of Biology and Investigator in The Picower Institute for Learning and Memory, seeks to understand the ways our nervous systems detect and respond to stimuli in health and disease.
Q: You’re interested in interoceptive biology. What makes the nervous system of mice a good model for doing that?
A: Our flagship system is the mammalian airway. We use a mouse model and modern molecular neuroscience tools to manipulate various neural pathways and observe what the effects are on respiratory function and animal health.
Neuroscience and mouse work have a reputation for being a little challenging and intense, but I think this is also where we can ask really important questions that are useful for our everyday lives — and the only place where we can fully recapitulate the complexity of nervous system signaling all the way down to our organs, back to our brain, and back to our organs.
It’s a very fun place to do science with lots of open questions.
One of the core discoveries from my postdoctoral work was focusing on the vagus nerve as a major body-to-brain conduit, as it innervates our lungs, heart and gastrointestinal tract. We found that there were about 40 different subtypes of sensory neurons within this small nerve, which is really a remarkable amount of diversity and reflects the massive sensory space within the body. About a dozen of those vagal neurons project to the airways.
We identified a rare neuron type specifically responsible for triggering protective responses like coughing when water or acid entered the airway. We also discovered a separate population of neurons that make us feel and act sick when we get a flu infection. The field now knows what four to five vagal populations of neurons are actually sensing in the airways, but the remaining populations are still a mystery to us; we don’t know what those populations of sensory neurons are detecting, what their anatomy is, and what reflex effects those neurons are evoking.
Looking ahead, there are many exciting directions for the interoceptive biology field. For example, there’s been a lot of focus on characterizing the circuits underlying acute motor reflexes, like rapid responses to visceral stimuli on the timescale of minutes to hours. But we don’t have a lot of information about what happens when these circuits are activated over long periods of time. For example, respiratory tract infections often last for weeks or longer. We know that the airways undergo changes in composition when they’re exposed to different types of infection or stress to better accommodate future threats. One of the hypotheses we’re testing is that chronically activating neural circuits may drive changes in organ composition. We have this idea, which we’re calling reflexive remodeling: neurons may be communicating with stem cells and progenitor cells in the periphery to drive adaptive remodeling responses.
We have the genetic, molecular and circuit scale tools to explore this phenomenon in mice. In parallel, we’re also setting up some in vitro models of the mouse airway mucosa to expedite receptor screening and to explore basic mechanisms of neuron-epithelium crosstalk. We hope this will inform our understanding of how the airway surface senses and responds to different types of irritants or damage.
Q: Why is understanding the peripheral nervous system important, and what parts of your background are you drawing on for your current research?
A: The lab focuses on really trying to explore the body-brain connection.
People often think that our mind exists in a vacuum, but in reality, our nervous system is heavily integrated with the rest of the body, and those neural interfaces are important, both for taking information from our body or environment and turning it into an internal representation of the world, and, in reverse, being able to process that information and being able to enact changes throughout the body. That includes things like autonomic reflexes, basic functions of the body like breathing, blood-gas regulation, digestion, and heart rate.
I’ve integrated both my graduate training and postdoctoral training into thinking about biology across multiple scales.
Graduate school for me was quite focused on deep molecular mechanism questions, particularly gene regulation, so I feel like that has been very useful for me in my general approach to neuroscience because I take a very molecular angle to all of this.
It also showed me the power of in vitro models as reductionist tools to explore fundamental aspects of cell biology. During my postdoc, I focused on larger, emergent phenotypes. We were able to manipulate specific circuits and see very impressive behavioral responses in animals. You could stimulate about 100 neurons in a mouse and see that their breathing would just stop until you remove the stimulation, and then the breathing would return to normal.
Both of those experiences inform how we approach a problem in my research. We need to understand how these circuits work, not just their connectivity at the anatomical level but what is driving their changes in sensitivity over time, the receptor expression programs that affect how they sense and signal, how these circuits emerge during development, and their gene expression.
There are still so many foundational questions that haven’t been answered that there’s enough to do in the mouse for quite some time.
Q: This all sounds fascinating. Where does it lead?
A: Human health has been my north star for a long time and I’ve taken a long, wandering path to find particular areas where I can scratch whatever intellectual itch that I have.
I originally thought I would be a doctor and then realized that I felt like I could have a more lasting impact by discovering fundamental truths about how our bodies work. I think there are a number of chronic diseases in which autonomic imbalance is actually a huge clinical component of the disorder.
We have a lot of interest in some of these very common airway remodeling diseases, like chronic obstructive pulmonary disorder—COPD—asthma, and potentially lung cancer. We want to ask questions like how autonomic circuits are altered in disease contexts, and when neurons actually drive features of disease.
Perhaps this research will help us come up with better molecular, cellular or tissue engineering approaches to improve the outcomes for a variety of autonomic diseases.
It’s very easy for me to imagine how one day not too far from now we can turn these findings into something actionable for human health.