Eva Frederick | Whitehead Institute
August 23, 2021
More than 10 percent of our genome is made up of repetitive, seemingly nonsensical stretches of genetic material called satellite DNA that do not code for any proteins. In the past, some scientists have referred to this DNA as “genomic junk.”
Over a series of papers spanning several years, however, Whitehead Institute Member Yukiko Yamashita and colleagues have made the case that satellite DNA is not junk, but instead has an essential role in the cell: it works with cellular proteins to keep all of a cell’s individual chromosomes together in a single nucleus.
Now, in the latest installment of their work, published online July 24 in the journal Molecular Biology and Evolution, Yamashita and former postdoctoral fellow Madhav Jagannathan, currently an assistant professor at ETH Zurich, Switzerland, take these studies a step further, proposing that the system of chromosomal organization made possible by satellite DNA is one reason that organisms from different species cannot produce viable offspring.
“Seven or eight years ago when we decided we wanted to study satellite DNA, we had zero plans to study evolution,” said Yamashita, who is also a professor of biology at the Massachusetts Institute of Technology and an investigator with the Howard Hughes Medical Institute. “This is one very fun part of doing science: when you don’t have a preconceived idea, and you just follow the lead until you bump into something completely unexpected.”
The origin of species: DNA edition
Researchers have known for years that satellite DNA is highly variable between species. “If you look at the chimpanzee genome and the human genome, the protein coding regions are, like, 98 percent, 99 percent identical,” she says. “But the junk DNA part is very, very different.”
“These are about the most rapidly evolving sequences in the genome, but the prior perspective has been, ‘Well, these are junk sequences, who cares if your junk is different from mine?’” said Jagannathan.
But as they were investigating the importance of satellite DNA for fertility and survival in pure species, Yamashita and Jagannathan had their first hint that these repetitive sequences might play a role in speciation.
When the researchers deleted a protein called Prod that binds to a specific satellite DNA sequence in the fruit fly Drosophila melanogaster, the flies’ chromosomes scattered outside of the nucleus into tiny globs of cellular material called micronuclei, and the flies died. “But we realized at this point that this [piece of] satellite DNA that was bound by the Prod protein was completely missing in the nearest relatives of Drosophila melanogaster,” Jagannathan said. “It completely doesn’t exist. So that’s an interesting little problem.”
If that piece of satellite DNA was essential for survival in one species but missing from another, it could imply that the two species of flies had evolved different satellite DNA sequences for the same role over time. And since satellite DNA played a role in keeping all the chromosomes together, Yamashita and Jagannathan wondered whether these evolved differences could be one reason different species are reproductively incompatible.
“After we realized the function [of satellite DNA in the cell], the fact that satellite DNA is quite different between species really hit like lightning,” Yamashita said. “All of a sudden, it became a completely different investigation.”
A tale of two fruit fly species
To study how satellite DNA differences might underlie reproductive incompatibility, the researchers decided to focus on two branches of the fruit fly family tree: the classic lab model Drosophila melanogaster, and its closest relative, Drosophila simulans. These two species diverged from each other two to three million years ago.
Researchers can breed a Drosophila melanogaster female to a Drosophila simulans male, “but [the cross] generates very unhappy offspring,” Yamashita said. “Either they’re sterile or they die.”
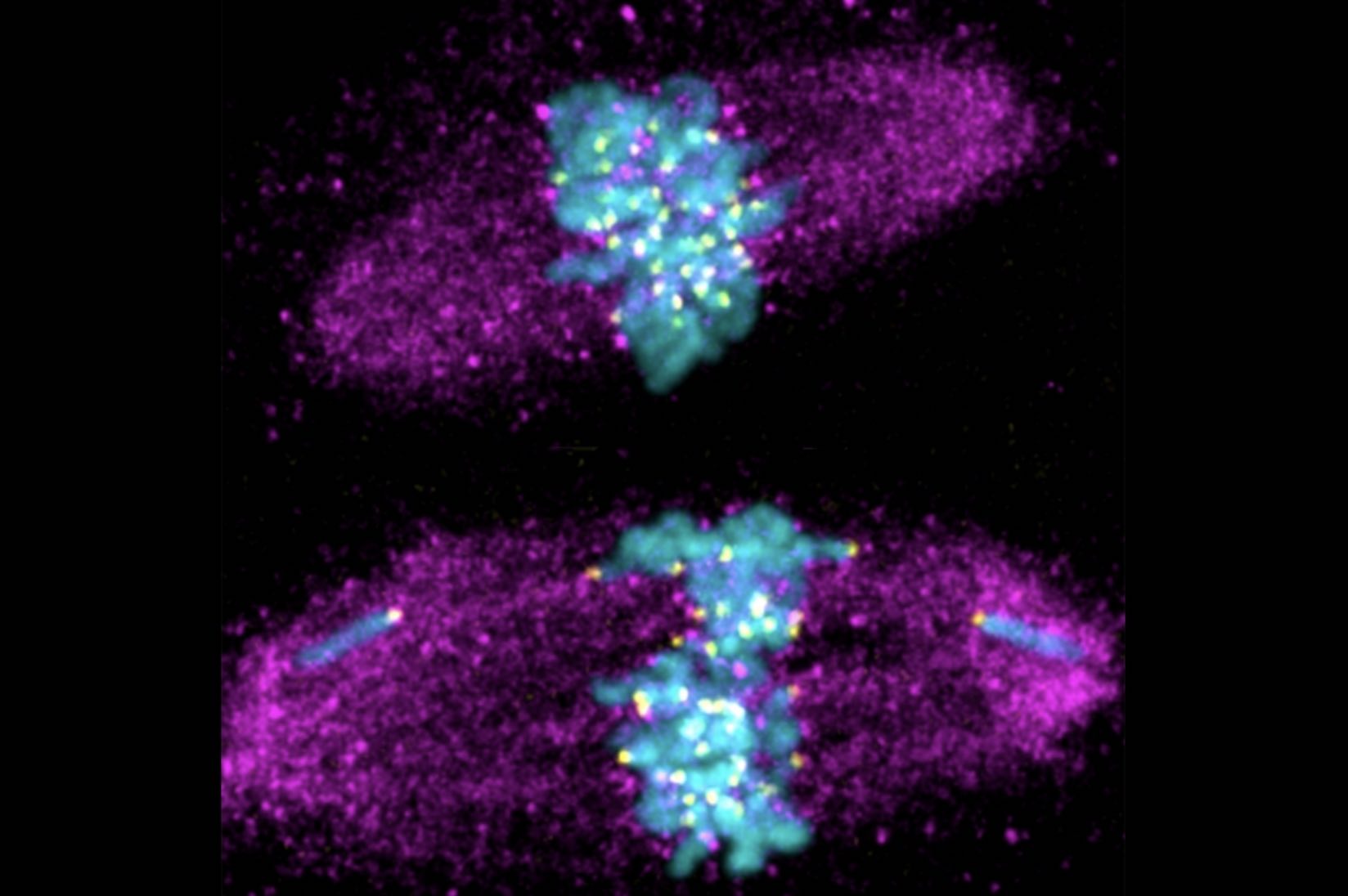
Yamashita and Jagannathan bred the flies together, then studied the tissues of the offspring to see what was leading these “unhappy” hybrids to drop like flies. Right away they noticed something interesting: “When we looked at those hybrid tissues, it was very clear that their phenotype was exactly the same as if you had disrupted the satellite DNA [-mediated chromosomal organization] of a pure species,” Yamashita said. “The chromosomes were scattered, and not encapsulated in a single nucleus.”
Furthermore, the researchers could create a healthy hybrid fly by mutating certain genes in the parent flies called “hybrid incompatibility genes,” which have been shown to localize to satellite DNA in the cells of pure species. Via these experiments, the researchers were able to demonstrate how these genes affect chromosomal packaging in hybrids, and pinpoint the cellular phenotypes associated with them for the first time. “I think for me, that is probably the most critical part of this paper,” Jagannathan said.
Taken together, these findings suggest that because satellite DNA mutates relatively frequently, the proteins that bind the satellite DNA and keep chromosomes together must evolve to keep up, leading each species to develop their own “strategy” for working with the satellite DNA. When two organisms with different strategies interbreed, a clash occurs, leading the chromosomes to scatter outside of the nucleus.
In future studies, Yamashita and Jagannathan hope to put their model to the ultimate test: if they can design a protein that can bind the satellite DNA of two different species and hold the chromosomes together, they could theoretically ‘rescue’ a doomed hybrid, allowing it to survive and produce viable offspring.
This feat of bioengineering is likely years off. “Right now it’s just a pure conceptual thing,” Yamashita said. “In doing this tinkering, there’s probably a lot of specifics that will have to be solved.”
For now, the researchers plan to continue investigating the roles of satellite DNA in the cell, armed with their new knowledge of the part it plays in speciation. “To me, the surprising part of this paper is that our hypothesis was correct,” Jagannathan said. “I mean, in retrospect, there are so many ways things could have been inconsistent with what we hypothesized, so it’s kind of amazing that we’ve sort of been able to chart a clear path from start to finish.”