
Ferroptosis is an iron-dependent form of cell death with profound implications in human health and disease. In the context of cancer, the use of ferroptosis inducers to target subpopulations of highly metastatic and therapy-resistant cancer cells has garnered much excitement over the last few years. However, to gain a comprehensive understanding of the full therapeutic potential of ferroptosis, our research focuses on (i) uncovering the molecular factors affecting ferroptosis susceptibility, (ii) studying its impact on the tumor microenvironment, and (iii) developing innovative ways to modulate ferroptosis resistance in vivo. We employ a multidisciplinary approach, combining functional genomics, metabolomics, bioengineering, and a range of in vitro and in vivo models to advance our understanding in this domain and to translate our findings into effective therapies.
Germline stem cells are the pool of stem cells capable of becoming eggs or sperm. They divide asymmetrically, such that one of the cells resulting from a division is another stem cell and the other is a differentiated cell, which has progressed one step further down the path towards becoming an egg or sperm. Researchers have thought that this asymmetrical division served to replenish the pool of stem cells—making sperm or eggs, but also making more stem cells to produce future sperm or eggs. However, the germline has another way to replenish itself: cells that have differentiated only one or two steps down the path to becoming eggs or sperm are capable of reverting into stem cells. Why, then, do stem cells divide asymmetrically?
New research from Whitehead Institute Member Yukiko Yamashita, who is also a professor of biology at the Massachusetts Institute of Technology and an HHMI Investigator, and former postdoc in her lab Jonathan Nelson shows that asymmetrical division in germline stem cells serves a different but equally important purpose in male fruit flies (Drosophila melanogaster), a common model animal for germline research. The work, published in the journal Proceedings of the National Academy of Sciences (PNAS) on November 13, suggests that in flies, germline stem cells divide asymmetrically in order to unequally split a certain kind of DNA, called ribosomal DNA (rDNA), between the two dividing cells and then keep the cell with more rDNA in the stem cell pool. This is necessary in order to keep the germline viable over generations of cell divisions, and so to keep individual flies fertile and capable of reproduction. The researchers show that only germline stem cells, and not other types of germ cells, drive this process, and explain why stem cells’ asymmetric divisions make them uniquely suited to maintaining rDNA.
Ribosomal DNA is critical to maintain in the germline because it contains the instructions for making a major part of ribosomes, the cellular machines that build proteins from genetic instructions. Proteins are the main workhorses of the cell, and so cells need to make many ribosomes in order to build all of the proteins that they need. Consequently, rDNA exists as many copies repeated in a row of the code for components of the ribosome. All of these repeats make it easy for the cell to mass produce ribosomes, but they also come with a risk: repetitive DNA is prone to losing repeats during cell division. When the cell’s rDNA is copied, it’s easy for a few of the many identical repeats to get cut out, so that the resulting copy of the genome has fewer rDNA repeats than the original.
Most cells can afford to lose a few rDNA repeats without too many negative effects, but the germline cannot. Whereas other cells die with the body they are in, germ cells produce eggs and sperm that will form a new body, which produces new germ cells, and so on. The germ cell lineage is effectively immortal. Over the course of its endless cycle of cell division, the loss of rDNA repeats would add up until the cells became dysfunctional and then died. This would make the individual bearing those germ cells infertile, and so cause their lineage to go extinct.
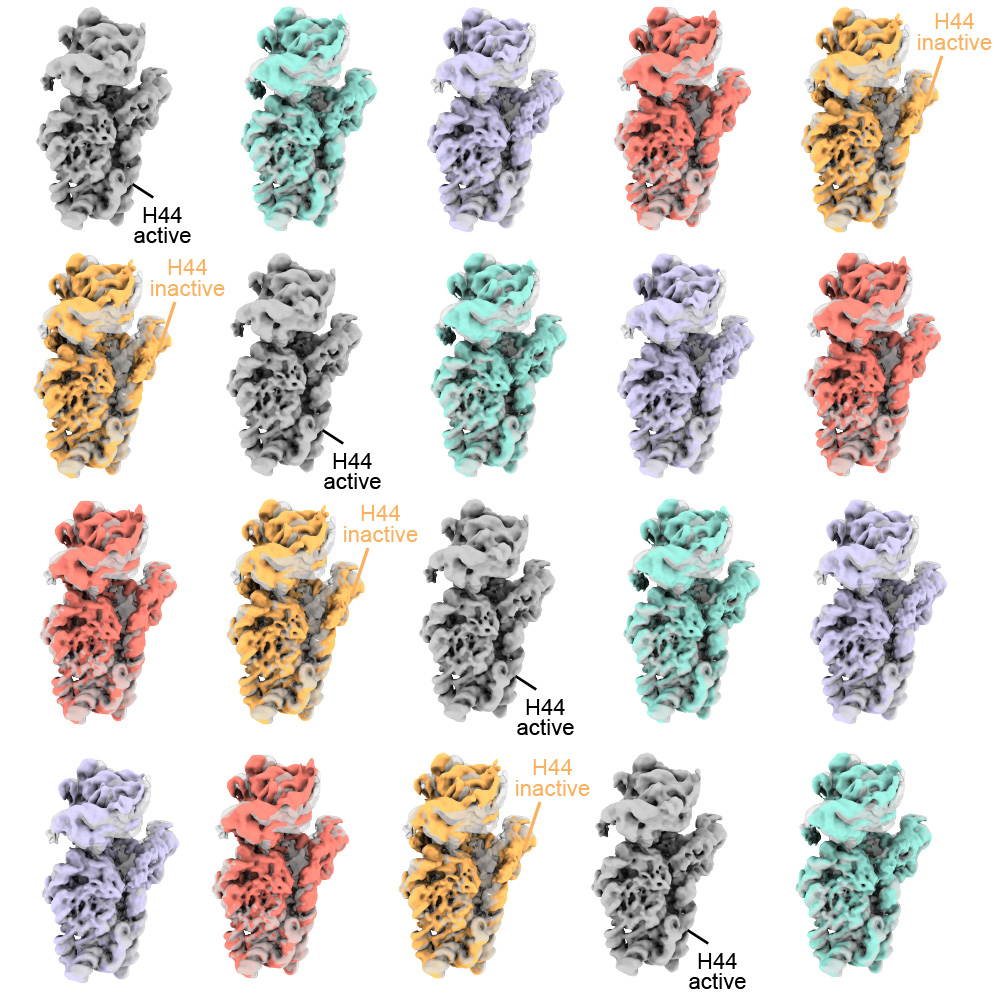
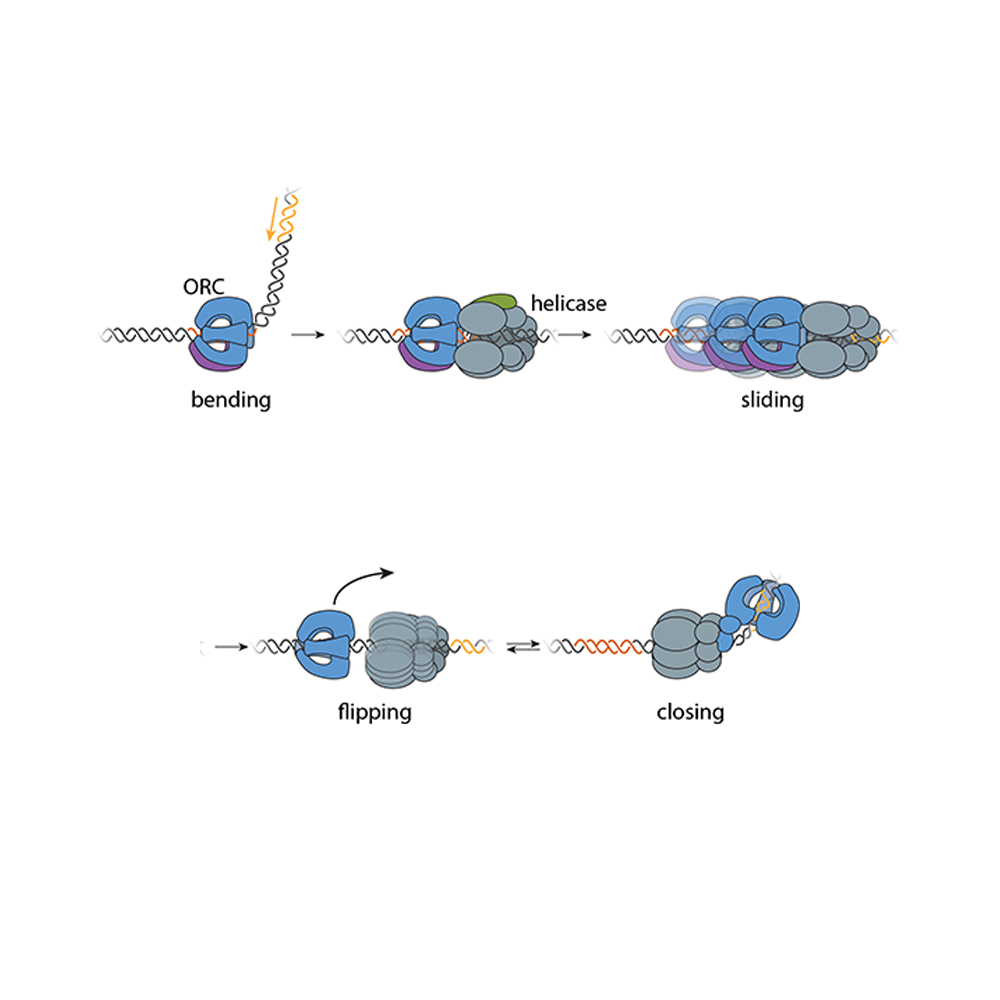
Researchers have known that germ cells have some way to regain rDNA repeats when the number gets too low—if germ cells couldn’t do this, none of us would exist—but the details of how cells achieve this have been largely mysterious. One proposed model was that when a germ cell divides, sometimes it might divide up its rDNA unequally between the two resulting cells, so that one cell would gain rDNA repeats. Yamashita and Nelson have previously found evidence that this model is correct, and they discovered some of the specific mechanisms that enable it to happen. In a 2023 PNAS paper, the researchers showed that a retrotransposon, a “selfish” genetic element whose function is to make more copies of itself, actually helps germ cells maintain rDNA. During cell division, the retrotransposon R2 slices open one copy of the chromosome containing rDNA in its quest to insert extra copies of itself into the genome. The cell tries to repair the break using the copy on the other intact chromosome, but the tricky nature of repetitive DNA can cause the cell to lose its place, so that it stitches a stretch of rDNA repeats from one copy of the chromosome into the other copy instead.
Through this process, the germline can boost the level of rDNA in a cell—but only by as much as another cell loses. How does this win-lose exchange lead to an overall increase in rDNA levels across the germline cell population to compensate for lost rDNA? In this latest work, Yamashita and Nelson show through mathematical modeling that in cells that divide symmetrically, it would not. Gains and losses in rDNA through this form of exchange would occur essentially at random and cancel each other out over time.
Now consider an asymmetric division. After a germline stem cell divides, the cell that differentiates will go through a few more divisions and ultimately create a specific number of sperm cells–the number happens to be sixty-four. If this daughter cell gets the chromosome with more rDNA repeats, then that would lead to sixty-four sperm with more rDNA repeats—but that would be it, as the sperm have exited the pool of replicating germline stem cells.
However, the daughter cell that remains a germline stem cell will divide again to create a differentiated cell (which will become sixty-four sperm) and another stem cell, which will divide again, leading to another sixty-four sperm and another stem cell—and so on. All of these cells, including many sperm, would inherit the higher number of rDNA repeats. Furthermore, at each division, there would be an opportunity for another unequal split of rDNA. As long as the stem cell always gets the boost in rDNA, then the cumulative number of rDNA repeats would keep growing in the overall population over time—and Yamashita and colleagues’ past work shows that the germline can ensure this. A 2022 Science Advances paper from Yamashita and then-postdoc in her lab George Watase showed that when a germline stem cell divides, the DNA strand with more rDNA repeats is tagged with a protein that the researchers named Indra, which helps mark it to stay in the daughter cell that will become another stem cell. Yamashita and Nelson’s new paper includes mathematical modeling by second author Tomohiro Kumon, a postdoc in Yamashita’s lab, that proves that this is not only sufficient to restore the level of rDNA repeats over time, but that it is the most effective and efficient way for the germline to do so.
“There was this problem with the unequal exchange model of rescuing rDNA, because every cell that gained rDNA did so at the expense of another that was losing it,” Nelson says. “What we show here is that the reason why there’s a bias towards gain in the germline is because this process is happening within these asymmetrically dividing germline stem cells that can gain and gain and gain, while the cells that lose rDNA exit the cycle and so have a limited effect.”
The researchers complemented their mathematical modeling with evidence that the process to increase rDNA repeats occurs primarily or solely in germline stem cells. They found that when the number of rDNA repeats got low enough, then expression of R2 and the presence of double-stranded DNA breaks both increased in germline stem cells, but not significantly in other germ cell types.
Yamashita and Nelson propose that the different cell types in the germline take on different functions to create a pipeline for maximizing the health of future sperm. Germ cells that are one or two steps down the path of differentiation from stem cells are essentially identical to them, to the point that they can be difficult to tell apart in testing, but they divide symmetrically. They are also much more sensitive to DNA damage; the researchers found that R2 exposure kills these cells.
Germline stem cells, with their asymmetrical division and ability to tolerate R2 expression, serve to restore rDNA levels when they get too low. Then the differentiated germ cells serve to weed out mutations—including those introduced during R2 expression in the earlier stem cell stage—by killing off cells with DNA damage. The different strengths of the different types of germ cells creates an effective pipeline to produce the largest number of sperm cells with high rDNA repeat number and low DNA damage.
Eventually, this new understanding of the details of how cells maintain their rDNA could lead to medical therapies. For example, cancer cells are, like germ cells, an essentially immortal cell line, and so must have a way to maintain their rDNA. If researchers could someday find a way to prevent them from doing so, that could be a good treatment strategy. The work also may have implications for research on aging, as rDNA decreases with age in other cell types. In the meantime, Yamashita and Nelson are excited to have solved several long-standing mysteries in their field, including how germ cells can restore rDNA at a population level when each division creates an equal loss and gain of rDNA, and why germline stem cells divide asymmetrically.
“Typically, when you publish a paper, you feel like you’ve fit two puzzle pieces together, but in this case, I feel like we fit a bunch of puzzle pieces together,” Yamashita says. “It’s been immensely satisfying to find answers to multiple questions and see how they all fit together to explain the mechanisms of this process that’s necessary for germline immortality.”



Sipping a beer on an early autumn evening, one might not consider that humans and yeast have been inextricably linked for thousands of years; winemaking, baking, and brewing all depend on budding yeast. Outside of baking and fermentation, researchers also use Saccharomyces cerevisiae, classified as a fungus, to study fundamental questions of cell biology.
Budding yeast gets its name from the way it multiplies. A daughter cell forms first as a swelling, protruding growth on the mother cell. The daughter cell projects further and further from the mother cell until it detaches as an independent yeast cell.
How do cells decide on a front and back? How do cells decode concentration gradients of chemical signals to orient in useful directions, or sense and navigate around physical obstacles? New Department of Biology faculty member Daniel “Danny” Lew uses the model yeast S. cerevisiae, and a non-model yeast with an unusual pattern of cell division, to explore these questions.
Q: Why is it useful to study yeast, and how do you approach the questions you hope to answer?
A: Humans and yeast are descended from a common ancestor, and some molecular mechanisms developed by that ancestor have been around for so long that yeast and mammals often use the same mechanisms. Many cells develop a front and migrate or grow in a particular direction, like the axons in our nervous system, using similar molecular mechanisms to those of yeast cells orienting growth towards the bud.
When I started my lab, I was working on cell cycle control, but I’ve always been interested in morphogenesis and the cell biology of how cells change shape and decide to do different things with different parts of themselves. Those mechanisms turn out to be conserved between yeast and humans.
But some things are very different about fungal and animal cells. One of the differences is the cell wall and what fungal cells have to do to deal with the fact that they have a cell wall.
Fungi are inflated by turgor pressure, which pushes their membranes against the rigid cell wall. This means they’ll die if there is any hole in the cell wall, which would be expected to happen often as cells remodel the wall in order to grow. We’re interested in understanding how fungi sense when any weak spots appear in the wall and repair them before those weak spots become dangerous.
Yeast cells, like most fungi, also mate by fusing with a partner. To succeed, they must do the most dangerous thing in the fungal life cycle: get rid of the cell wall at the point of contact to allow fusion. That means they must be precise about where and when they remove the wall. We’re fascinated to understand how they know it is safe to remove the wall there, and nowhere else.
We take an interdisciplinary approach. We’ve used genetics, biochemistry, cell biology, and computational biology to try and solve problems in the past. There’s a natural progression: observation and genetic approaches tend to be the first line of attack when you know nothing about how something works. As you learn more, you need biochemical approaches and, eventually, computational approaches to understand exactly what mechanism you’re looking at.
I’m also passionate about mentoring, and I love working with trainees and getting them fascinated by the same problems that fascinate me. I’m looking to work with curious trainees who love addressing fundamental problems.
Q: How does yeast decide to orient a certain way — toward a mating partner, for example?
A: We are still working on questions of how cells analyze the surrounding environment to pick a direction. Yeast cells have receptors that sense pheromones that a mating partner releases. What is amazing about that is that these cells are incredibly small, and pheromones are released by several potential partners in the neighborhood. That means yeast cells must interpret a very confusing landscape of pheromone concentrations. It’s not apparent how they manage to orient accurately toward a single partner.
That got me interested in related questions. Suppose the cell is oriented toward something that isn’t a mating partner. The cell seems to recognize that there’s an obstacle in the way, and it can change direction to go around that obstacle. This is how fungi get so good at growing into things that look very solid, like wood, and some fungi can even penetrate Kevlar vests.
If they recognize an obstacle, they have to change directions and go around it. If they recognize a mating partner, they have to stick with that direction and allow the cell wall to get degraded. How do they know they’ve hit an obstacle? How do they know a mating partner is different from an obstacle? These are the questions we’d like to understand.
Q: For the last couple of years, you’ve also been studying a budding yeast that forms multiple buds when it reproduces instead of just one. How did you come across it, and what questions are you hoping to explore?
A: I spent several years trying to figure out why most yeasts make one bud and only one bud, which I think is related to the question of why migrating cells make one and only one front. We had what we thought was a persuasive answer to that, so seeing a yeast completely disobey that and make as many buds as it felt like was a shock, which got me intrigued.
We started working on it because my colleague, Amy Gladfelter, had sampled the waters around Woods Hole, Massachusetts. When she saw this specimen under a microscope, she immediately called me and said, “You have to look at this.”
A question we’re very intrigued by is if the cell makes five, seven, or 12 buds simultaneously, how do they divide the mother cell’s material and growth capacity five, seven, or 12 ways? It looks like all of the buds grow at the same rate and reach about the same size. One of our short-term goals is to check whether all the buds really get to exactly the same size or whether they are born unequal.
And we’re interested in more than just growth rate. What about organelles? Do you give each bud the same number of mitochondria, nuclei, peroxisomes, and vacuoles? That question will inevitably lead to follow-up questions. If each bud has the same number of mitochondria, how does the cell measure mitochondrial inheritance to do that? If they don’t have the same amount, then buds are each born with a different complement and ratio of organelles. What happens to buds if they have very different numbers of organelles?
As far as we can tell, every bud gets at least one nucleus. How the cell ensures that each bud gets a nucleus is a question we’d also very much like to understand.
We have molecular candidates because we know a lot about how model yeasts deliver nuclei, organelles, and growth materials from the mother to the single bud. We can mutate candidate genes and see if similar molecular pathways are involved in the multi-budding yeast and, if so, how they are working.
It turns out that this unconventional yeast has yet to be studied from the point of view of basic cell biology. The other thing that intrigues me is that it’s a poly-extremophile. This yeast can survive under many rather harsh conditions: it’s been isolated in Antarctica, from jet engines, from all kinds of plants, and of course from the ocean as well. An advantage of working with something so ubiquitous is we already know it’s not toxic to us under almost any circumstances. We come into contact with it all the time. If we learn enough about its cell biology to begin to manipulate it, then there are many potential applications, from human health to agriculture.
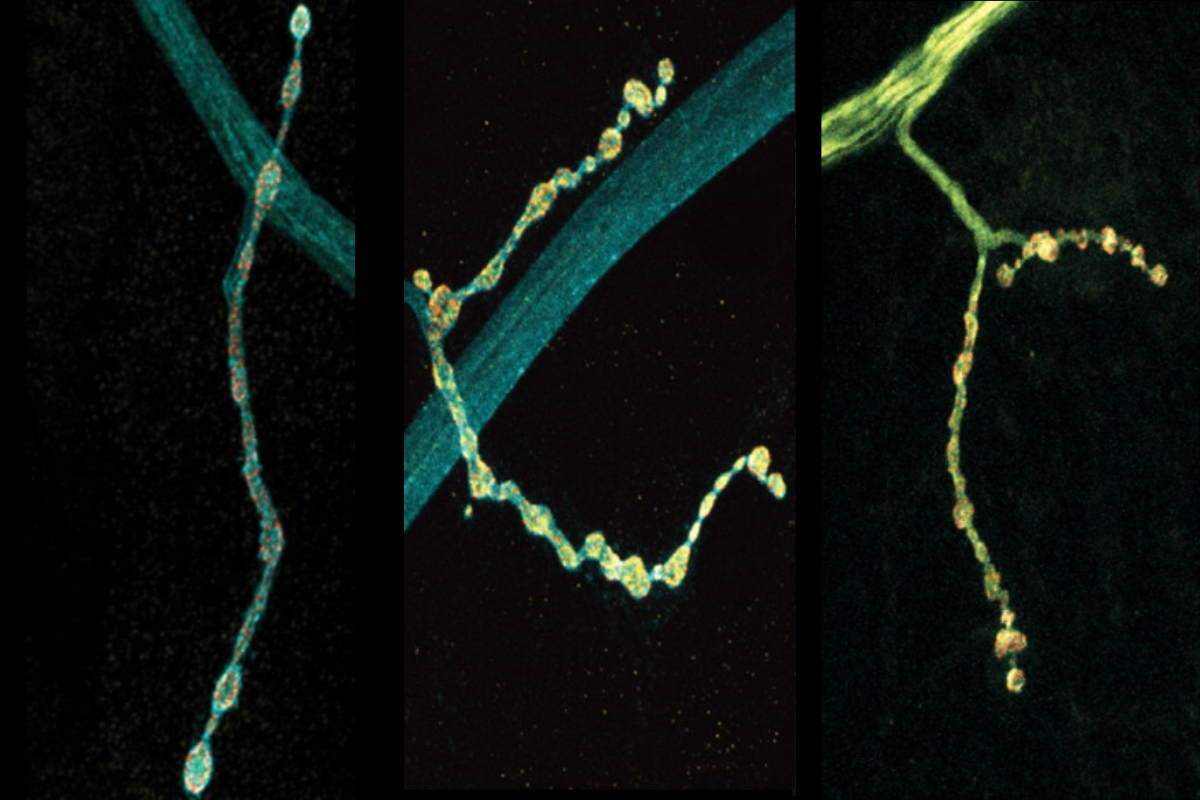
Neurons are talkers. They each communicate with fellow neurons, muscles, or other cells by releasing neurotransmitter chemicals at “synapse” junctions, ultimately producing functions ranging from emotions to motions. But even neurons of the exact same type can vary in their conversational style. A new open-access study in Cell Reports by neurobiologists at The Picower Institute for Learning and Memory highlights a molecular mechanism that might help account for the nuanced diversity of neural discourse.
The scientists made their findings in neurons that control muscles in Drosophila fruit flies. These cells are models in neuroscience because they exhibit many fundamental properties common to neurons in people and other animals, including communication via the release of the neurotransmitter glutamate. In the lab of Troy Littleton, Menicon Professor in MIT’s departments of Biology and Brain and Cognitive Sciences, which studies how neurons regulate this critical process, researchers frequently see that individual neurons vary in their release patterns. Some “talk” more than others.
In more than a decade of studies, Littleton’s lab has shown that a protein called complexin has the job of restraining spontaneous glutamate chatter. It clamps down on fusion of glutamate-filled vesicles at the synaptic membrane to preserve a supply of the neurotransmitter for when the neuron needs it for a functional reason, for instance to simulate a muscle to move. The lab’s studies have identified two different kinds of complexin in flies (mammals have four) and showed that the clamping effectiveness of the rare but potent 7B splice form is regulated by a molecular process called phosphorylation. How the much more abundant 7A version is regulated was not known, but scientists had shown that the RNA transcribed from DNA that instructs the formation of the protein is sometimes edited in the cell by an enzyme called ADAR.
In the new study from Littleton’s team, led by Elizabeth Brija PhD ’23, the lab investigated whether RNA editing of complexin 7A affects how it regulates glutamate release. What she discovered was surprising. Not only does RNA editing of complexin 7A have a significant impact on how well the protein prevents glutamate release, but also this can vary widely among individual neurons because they can stochastically mix and match up to eight different editions of the protein. Some edits were much more common than others on average, but 96 percent of the 200 neurons the team examined had at least some editing, which affected the structure of an end of the protein called its C-terminus. Experiments to test some of the consequences of this structural variation showed that different complexin 7A edits can dramatically affect the level of electrical current measurable at different synapses. That varying level of activity can also affect the growth of the synapses the neurons make with muscle. RNA editing of the protein might therefore endow each neuron with fine degrees of communication control.
“What this offers the nervous system is that you can take the same transcriptome and by alternatively editing various RNA transcripts, these neurons will behave differently,” Littleton says.
Moreover, Littleton and Brija’s team found that other key proteins involved in synaptic glutamate release, such as synapsin and Syx1A, are also sometimes edited at quite different levels among the same population of neurons. This suggests that other aspects of synaptic communication might also be tunable.
“Such a mechanism would be a robust way to change multiple features of neuronal output,” Brija, Littleton, and colleagues wrote.
The team tracked the different editing levels by meticulously extracting and sequencing RNA from the nuclei and cell bodies of 200 motor neurons. The work yielded a rich enough dataset to show that any of three adenosine nucleotides encoding two amino acids in the C-terminus could be swapped for another, yielding eight different editions of the protein. A slim majority of complexin 7A went unedited in the average neuron, while the seven edited versions composed the rest with widely varying degrees of frequency.
To investigate the functional consequences of some of the different editions, the team knocked out complexin and then “rescued” flies by adding back in unedited or two different edited versions. The experiments showed a stark contrast between the two edited proteins. One, which occurs more commonly, proved to be a less effective clamp than unedited complexin, barely preventing spontaneous glutamate release and upticks in electrical current. The other turned out to be more effective at clamping than the unedited version, keeping a tight lid on glutamate release and synaptic output. And while both of the edited versions showed a tendency to drift away from synapses and into the neuron’s axon, the long branch that extends from the cell body, the edition that clamped well prevented any overgrowth of synapses while the one that clamped poorly provided only a meager curb.
Because multiple editions are often present in neurons, Brija and the team did one more set of experiments in which they “rescued” complexin-less flies with a combination of unedited complexin and the weak-clamping edition. The result was a blend of the two: reduced spontaneous glutamate release than with just the weakly clamping edition alone. The findings suggest that not only does each edition potentially fine-tune glutamate release, but that combinations among them can act in a combinatorial fashion.
In addition to Brija and Littleton the paper’s other authors are Zhuo Guan and Suresh Jetti.
The National Institutes of Health, The JPB Foundation, and The Picower Institute for Learning and Memory supported the research.