New technique reveals many possible conformations that a protein may take.
Anne Trafton | MIT News Office
February 4, 2021
Cryo-electron microscopy (cryo-EM) allows scientists to produce high-resolution, three-dimensional images of tiny molecules such as proteins. This technique works best for imaging proteins that exist in only one conformation, but MIT researchers have now developed a machine-learning algorithm that helps them identify multiple possible structures that a protein can take.
Unlike AI techniques that aim to predict protein structure from sequence data alone, protein structure can also be experimentally determined using cryo-EM, which produces hundreds of thousands, or even millions, of two-dimensional images of protein samples frozen in a thin layer of ice. Computer algorithms then piece together these images, taken from different angles, into a three-dimensional representation of the protein in a process termed reconstruction.
In a Nature Methods paper, the MIT researchers report a new AI-based software for reconstructing multiple structures and motions of the imaged protein — a major goal in the protein science community. Instead of using the traditional representation of protein structure as electron-scattering intensities on a 3D lattice, which is impractical for modeling multiple structures, the researchers introduced a new neural network architecture that can efficiently generate the full ensemble of structures in a single model.
“With the broad representation power of neural networks, we can extract structural information from noisy images and visualize detailed movements of macromolecular machines,” says Ellen Zhong, an MIT graduate student and the lead author of the paper.
With their software, they discovered protein motions from imaging datasets where only a single static 3D structure was originally identified. They also visualized large-scale flexible motions of the spliceosome — a protein complex that coordinates the splicing of the protein coding sequences of transcribed RNA.
“Our idea was to try to use machine-learning techniques to better capture the underlying structural heterogeneity, and to allow us to inspect the variety of structural states that are present in a sample,” says Joseph Davis, the Whitehead Career Development Assistant Professor in MIT’s Department of Biology.
Davis and Bonnie Berger, the Simons Professor of Mathematics at MIT and head of the Computation and Biology group at the Computer Science and Artificial Intelligence Laboratory, are the senior authors of the study, which appears today in Nature Methods. MIT postdoc Tristan Bepler is also an author of the paper.
Visualizing a multistep process
The researchers demonstrated the utility of their new approach by analyzing structures that form during the process of assembling ribosomes — the cell organelles responsible for reading messenger RNA and translating it into proteins. Davis began studying the structure of ribosomes while a postdoc at the Scripps Research Institute. Ribosomes have two major subunits, each of which contains many individual proteins that are assembled in a multistep process.
To study the steps of ribosome assembly in detail, Davis stalled the process at different points and then took electron microscope images of the resulting structures. At some points, blocking assembly resulted in accumulation of just a single structure, suggesting that there is only one way for that step to occur. However, blocking other points resulted in many different structures, suggesting that the assembly could occur in a variety of ways.
Because some of these experiments generated so many different protein structures, traditional cryo-EM reconstruction tools did not work well to determine what those structures were.
“In general, it’s an extremely challenging problem to try to figure out how many states you have when you have a mixture of particles,” Davis says.
After starting his lab at MIT in 2017, he teamed up with Berger to use machine learning to develop a model that can use the two-dimensional images produced by cryo-EM to generate all of the three-dimensional structures found in the original sample.
In the new Nature Methods study, the researchers demonstrated the power of the technique by using it to identify a new ribosomal state that hadn’t been seen before. Previous studies had suggested that as a ribosome is assembled, large structural elements, which are akin to the foundation for a building, form first. Only after this foundation is formed are the “active sites” of the ribosome, which read messenger RNA and synthesize proteins, added to the structure.
In the new study, however, the researchers found that in a very small subset of ribosomes, about 1 percent, a structure that is normally added at the end actually appears before assembly of the foundation. To account for that, Davis hypothesizes that it might be too energetically expensive for cells to ensure that every single ribosome is assembled in the correct order.
“The cells are likely evolved to find a balance between what they can tolerate, which is maybe a small percentage of these types of potentially deleterious structures, and what it would cost to completely remove them from the assembly pathway,” he says.
Viral proteins
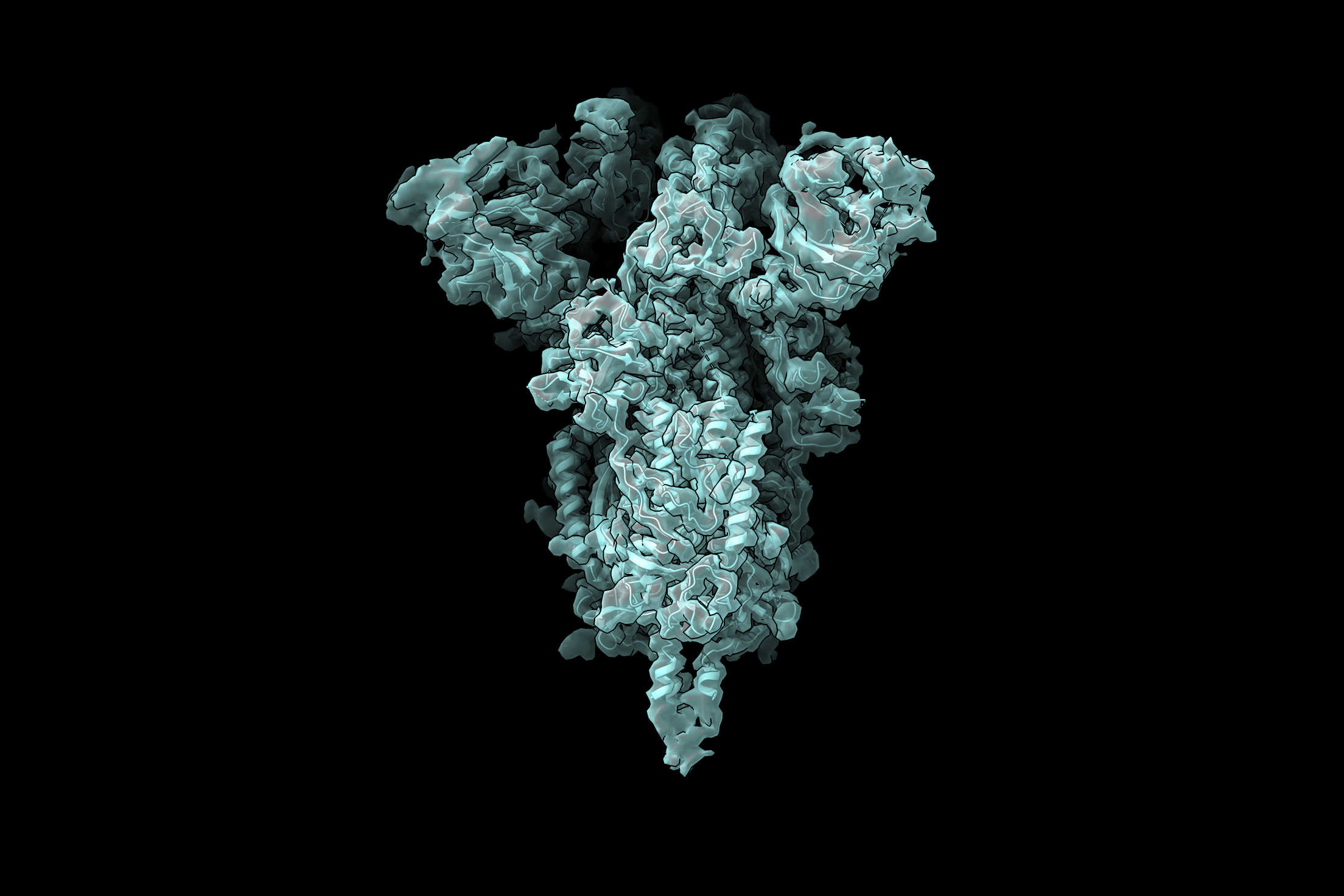
The researchers are now using this technique to study the coronavirus spike protein, which is the viral protein that binds to receptors on human cells and allows them to enter cells. The receptor binding domain (RBD) of the spike protein has three subunits, each of which can point either up or down.
“For me, watching the pandemic unfold over the past year has emphasized how important front-line antiviral drugs will be in battling similar viruses, which are likely to emerge in the future. As we start to think about how one might develop small molecule compounds to force all of the RBDs into the ‘down’ state so that they can’t interact with human cells, understanding exactly what the ‘up’ state looks like and how much conformational flexibility there is will be informative for drug design. We hope our new technique can reveal these sorts of structural details,” Davis says.
The research was funded by the National Science Foundation Graduate Research Fellowship Program, the National Institutes of Health, and the MIT Jameel Clinic for Machine Learning and Health. This work was supported by MIT Satori computation cluster hosted at the MGHPCC.