Study in worms reveals gene loss can lead to accumulation of waste products in cells.
Anne Trafton | MIT News Office
May 4, 2018
MIT biologists have discovered a function of a gene that is believed to account for up to 40 percent of all familial cases of amyotrophic lateral sclerosis (ALS). Studies of ALS patients have shown that an abnormally expanded region of DNA in a specific region of this gene can cause the disease.
In a study of the microscopic worm Caenorhabditis elegans, the researchers found that the gene has a key role in helping cells to remove waste products via structures known as lysosomes. When the gene is mutated, these unwanted substances build up inside cells. The researchers believe that if this also happens in neurons of human ALS patients, it could account for some of those patients’ symptoms.
“Our studies indicate what happens when the activities of such a gene are inhibited — defects in lysosomal function. Certain features of ALS are consistent with their being caused by defects in lysosomal function, such as inflammation,” says H. Robert Horvitz, the David H. Koch Professor of Biology at MIT, a member of the McGovern Institute for Brain Research and the Koch Institute for Integrative Cancer Research, and the senior author of the study.
Mutations in this gene, known as C9orf72, have also been linked to another neurodegenerative brain disorder known as frontotemporal dementia (FTD), which is estimated to affect about 60,000 people in the United States.
“ALS and FTD are now thought to be aspects of the same disease, with different presentations. There are genes that when mutated cause only ALS, and others that cause only FTD, but there are a number of other genes in which mutations can cause either ALS or FTD or a mixture of the two,” says Anna Corrionero, an MIT postdoc and the lead author of the paper, which appears in the May 3 issue of the journal Current Biology.
Genetic link
Scientists have identified dozens of genes linked to familial ALS, which occurs when two or more family members suffer from the disease. Doctors believe that genetics may also be a factor in nonfamilial cases of the disease, which are much more common, accounting for 90 percent of cases.
Of all ALS-linked mutations identified so far, the C9orf72 mutation is the most prevalent, and it is also found in about 25 percent of frontotemporal dementia patients. The MIT team set out to study the gene’s function in C. elegans, which has an equivalent gene known as alfa-1.
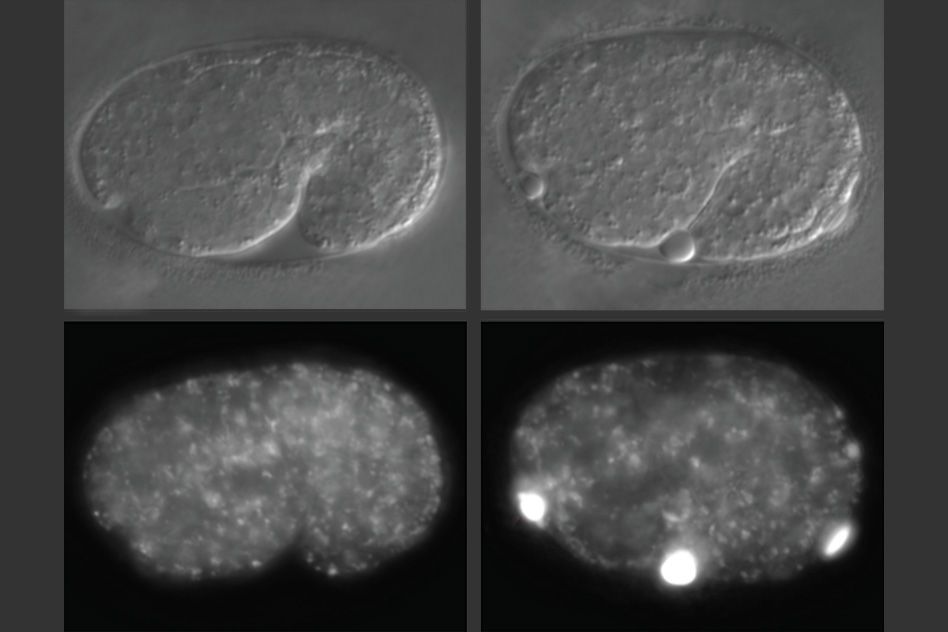
In studies of worms that lack alfa-1, the researchers discovered that defects became apparent early in embryonic development. C. elegans embryos have a yolk that helps to sustain them before they hatch, and in embryos missing alfa-1, the researchers found “blobs” of yolk floating in the fluid surrounding the embryos.
This led the researchers to discover that the gene mutation was affecting the lysosomal degradation of yolk once it is absorbed into the cells. Lysosomes, which also remove cellular waste products, are cell structures which carry enzymes that can break down many kinds of molecules.
When lysosomes degrade their contents — such as yolk — they are reformed into tubular structures that split, after which they are able to degrade other materials. The MIT team found that in cells with the alfa-1 mutation and impaired lysosomal degradation, lysosomes were unable to reform and could not be used again, disrupting the cell’s waste removal process.
“It seems that lysosomes do not reform as they should, and material accumulates in the cells,” Corrionero says.
For C. elegans embryos, that meant that they could not properly absorb the nutrients found in yolk, which made it harder for them to survive under starvation conditions. The embryos that did survive appeared to be normal, the researchers say.
Neuronal effects
The researchers were able to partially reverse the effects of alfa-1 loss in the C. elegans embryos by expressing the human protein encoded by the c9orf72 gene. “This suggests that the worm and human proteins are performing the same molecular function,” Corrionero says.
If loss of C9orf72 affects lysosome function in human neurons, it could lead to a slow, gradual buildup of waste products in those cells. ALS usually affects cells of the motor cortex, which controls movement, and motor neurons in the spinal cord, while frontotemporal dementia affects the frontal areas of the brain’s cortex.
“If you cannot degrade things properly in cells that live for very long periods of time, like neurons, that might well affect the survival of the cells and lead to disease,” Corrionero says.
Many pharmaceutical companies are now researching drugs that would block the expression of the mutant C9orf72. The new study suggests certain possible side effects to watch for in studies of such drugs.
“If you generate drugs that decrease c9orf72 expression, you might cause problems in lysosomal homeostasis,” Corrionero says. “In developing any drug, you have to be careful to watch for possible side effects. Our observations suggest some things to look for in studying drugs that inhibit C9orf72 in ALS/FTD patients.”
The research was funded by an EMBO postdoctoral fellowship, an ALS Therapy Alliance grant, a gift from Rose and Douglas Barnard ’79 to the McGovern Institute, and a gift from the Halis Family Foundation to the MIT Aging Brain Initiative.