Biologists discover the immune system can eliminate cells with too many or too few chromosomes.
Anne Trafton | MIT News Office
June 19, 2017
Most living cells have a defined number of chromosomes: Human cells, for example, have 23 pairs. As cells divide, they can make errors that lead to a gain or loss of chromosomes, which is usually very harmful.
For the first time, MIT biologists have now identified a mechanism that the immune system uses to eliminate these genetically imbalanced cells from the body. Almost immediately after gaining or losing chromosomes, cells send out signals that recruit immune cells called natural killer cells, which destroy the abnormal cells.
The findings raise the possibility of harnessing this system to kill cancer cells, which nearly always have too many or too few chromosomes.
“If we can re-activate this immune recognition system, that would be a really good way of getting rid of cancer cells,” says Angelika Amon, the Kathleen and Curtis Marble Professor in Cancer Research in MIT’s Department of Biology, a member of the Koch Institute for Integrative Cancer Research, and the senior author of the study.
Stefano Santaguida, a research scientist at the Koch Institute, is the lead author of the paper, which appears in the June 19 issue of Developmental Cell.
“A downward spiral”
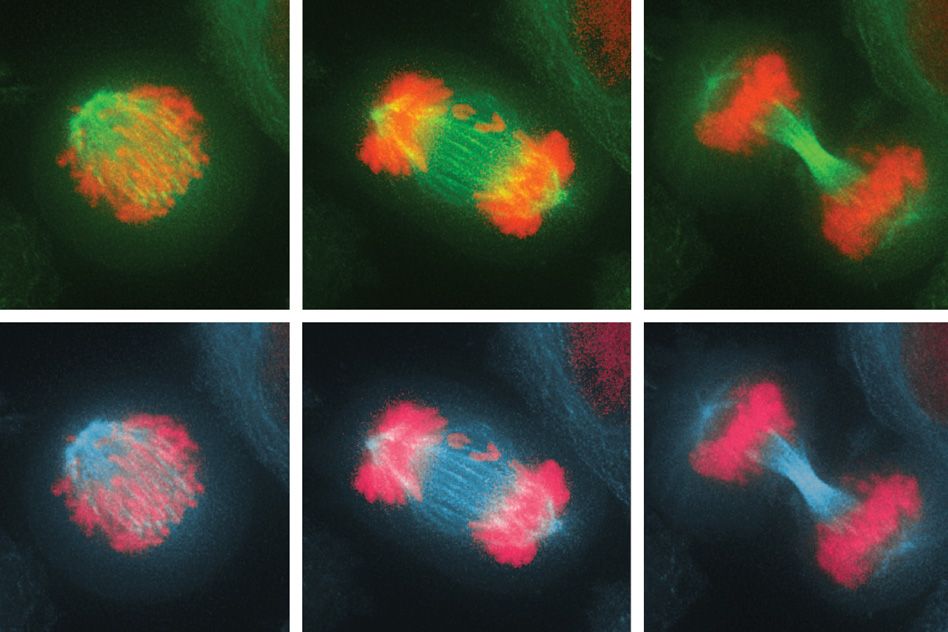
Before a cell divides, its chromosomes replicate and then line up in the middle of the cell. As the cell divides into two daughter cells, half of the chromosomes are pulled into each cell. If these chromosomes fail to separate properly, the process leads to an imbalanced number of chromosomes in the daughter cells — a state known as aneuploidy.
When aneuploidy occurs in embryonic cells, it is almost always fatal to the organism. For human embryos, extra copies of any chromosome are lethal, with the exceptions of chromosome 21, which produces Down syndrome; chromosomes 13 and 18, which lead to developmental disorders known as Patau and Edwards syndromes; and the X and Y sex chromosomes, extra copies of which may cause various disorders but are not usually lethal.
In recent years, Amon’s lab has been exploring an apparent paradox of aneuploidy: When normal adult cells become aneuploid, it impairs their ability to survive and proliferate; however, cancer cells, which are nearly all aneuploid, can grow uncontrollably.
“Aneuploidy is highly detrimental in most cells. However, aneuploidy is highly associated with cancer, which is characterized by upregulated growth. So, a very important question is: If aneuploidy hampers cell proliferation, why are the vast majority of tumors aneuploid?” Santaguida says.
To try to answer that question, the researchers wanted to find out more about how aneuploidy affects cells. Over the past few years, Santaguida and Amon have been studying what happens to cells immediately after they experience a mis-segregation of chromosomes, leading to imbalanced daughter cells.
In the new study, they investigated the effects of this imbalance on the cell division cycle by interfering with the process of proper chromosome attachment to the spindle, the structure that holds chromosomes in place at the cell’s equator before division. This interference leads some chromosomes to lag behind and get shuffled into the two daughter cells.
The researchers found that after the cells underwent their first division, in which some of the chromosomes were unevenly distributed, they soon initiated another cell division, which produced even more chromosome imbalance, as well as significant DNA damage. Eventually, the cells stopped dividing altogether.
“These cells are in a downward spiral where they start out with a little bit of genomic mess, and it just gets worse and worse,” Amon says.
“This paper very convincingly and clearly shows that when chromosomes are lost or gained, initially cells can’t tell if their chromosomes have mis-segregated,” says David Pellman, a professor of pediatric oncology at Dana-Farber Cancer Institute who was not involved in the study. “Instead, the imbalance of chromosomes leads to cellular defects and an imbalance of proteins and genes that can significantly disrupt DNA replication and cause further damage to the chromosomes.”
Targeting aneuploidy
As genetic errors accumulate, aneuploid cells eventually become too unstable to keep dividing. In this senescent state, they start producing inflammation-inducing molecules such as cytokines. When the researchers exposed these cells to immune cells called natural killer cells, the natural killer cells destroyed most of the aneuploid cells.
“For the first time, we are witnessing a mechanism that might provide a clearance of cells with imbalanced chromosome numbers,” Santaguida says.
In future studies, the researchers hope to determine more precisely how aneuploid cells attract natural killer cells, and to find out whether other immune cells are involved in clearing aneuploid cells. They would also like to figure out how tumor cells are able to evade this immune clearance, and whether it may be possible to restart the process in patients with cancer, since about 90 percent of solid tumors and 75 percent of blood cancers are aneuploid.
“At some point, cancer cells, which are highly aneuploid, are able to evade this immune surveillance,” Amon says. “We have really no understanding of how that works. If we can figure this out, that probably has tremendous therapeutic implications, given the fact that virtually all cancers are aneuploid.”
The research was funded, in part, by the National Institutes of Health, the Kathy and Curt Marble Cancer Research Fund, the American Italian Cancer Foundation, a Fellowship in Cancer Research from Marie Curie Actions, the Italian Association for Cancer Research, and a Koch Institute Quinquennial Cancer Research Fellowship.